Aranesp® (Darbepoetin Alfa) for Injection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Myelodysplastic Syndromes
MYELODYSPLASTIC SYNDROMES TREATMENT REGIMENS (Part 1 of 3) Clinical Trials: The National Comprehensive Cancer Network recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced healthcare team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are only provided to supplement the latest treatment strategies. These Guidelines are a work in progress that may be refined as often as new significant data becomes available. The NCCN Guidelines® are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines® is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. First-Line Treatment1 Note: All recommendations are Category 2A unless otherwise indicated. Relatively Lower-Risk Patientsa,b Symptomatic Anemia With del(5q) ± One Other Cytogenetic Abnormality (except those involving chromosome 7) REGIMEN DOSING Lenalidomide2-5,c Days 1–21: Lenalidomide 10mg orally once daily. Repeat cycle every 28 days (or 28 days monthly). Assess response 2–4 months after initiation of treatment. -

OMONTYS® Safely and Effectively
HIGHLIGHTS OF PRESCRIBING INFORMATION ---------------------DOSAGE FORMS AND STRENGTHS---------------------- These highlights do not include all the information needed to use Dosage Form Strengths OMONTYS® safely and effectively. See full prescribing information for OMONTYS. Single use vials 2 mg/0.5 mL, 3 mg/0.5 mL, (preservative-free) 4 mg/0.5 mL, 5 mg/0.5 mL, and OMONTYS® (peginesatide) Injection, 6 mg/0.5 mL for intravenous or subcutaneous use Single use pre-filled syringes 1 mg/0.5 mL, 2 mg/0.5 mL, Initial U.S. Approval: 2012 (preservative-free) 3 mg/0.5 mL, 4 mg/0.5 mL, 5 mg/0.5 mL, and 6 mg/0.5 mL WARNING: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL Multiple use vials 10 mg/mL and 20 mg/2 mL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, (with preservative) THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE -------------------------------CONTRAINDICATIONS------------------------------ See full prescribing information for complete boxed warning. Uncontrolled hypertension (4). Chronic Kidney Disease: In controlled trials, patients experienced greater risks for -----------------------WARNINGS AND PRECAUTIONS------------------------ death, serious adverse cardiovascular reactions, and stroke Increased Mortality, Myocardial Infarction, Stroke, and when administered erythropoiesis-stimulating agents (ESAs) Thromboembolism: Using ESAs to target a hemoglobin level of to target a hemoglobin level of greater than 11 g/dL (5.1). greater than 11 g/dL increases the risk of serious adverse No trial has identified a hemoglobin target level, ESA dose, or cardiovascular reactions and has not been shown to provide dosing strategy that does not increase these risks (5.1). additional benefits (5.1). -
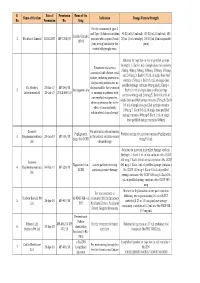
Final List of R-DNA Based Drugs Approved in the Country.Xlsx
S. Date of Permission Name of the Name of the firm Indication Dosage Form & Strength No. Permission No. Drug For the treatment of type -I and Type -II diabetes mellitus 40 IU/ml (10 ml vial), 100 IU/ml (10 ml vial), 100 Insulin Glargine 1 Wockhardt Limited 22.02.2007 MF-7206/07 patients who required basal IU/ml (3 ml cartridge), 100 IU/ml (3 ml disposable 100 IU (long acting) insulin for the pens) control of hyperglycemia. Solution for injection in vial or prefilled syringe Strength: 1. Each 1 mL of single dose vial contains Treatment of anaemia 25mcg, 40mcg, 60mcg, 100mcg, 200mcg, 300mcg associated with chronic renal and 500mcg 2. Each 0.75 mL of single dose vial failure, including patients on contains 150mcg 3. Each 0.3 mL of single dose dialysis and patients not on prefilled syringe contains 60mcg and 150mcg 4. Dr. Reddy's 23-Mar-10 MF-246/10 dialysis and for the treatment 2 Darbepoetin alfa Each 0.4 mL of single dose prefilled syringe Laboratories Ltd. 20-Jul-10 BULK-668/10 of anaemia in patients with contains 40mcg and 200mcg 5. Each 0.42 mL of non-myeloid malignancies, single dose prefilled syringe contains 25mcg 6. Each where anaemia is due to the 0.5 mL of single dose prefilled syringe contains effect of concomitantly 100mcg 7. Each 0.6 mL of single dose prefilled administered chemotherapy syringe contains 300mcg 8. Each 1 mL of single dose prefilled syringe contains 500mcg Gennova For prevention of neutropenia Pegfilgrastim Prefilled syringe for injection contains Pegfilgrastim 3 Biopharmaceuticals 29-Jan-10 MF-104/10 in the patient receiving cancer (peg-r-hu-GCSF) 6 mg/0.6 mL Ltd. -

Presentation Title
The COMMANDS trial: a phase 3 study of the efficacy and safety of luspatercept versus epoetin alfa for the treatment of anemia due to Revised International Prognostic Scoring System Very Low-, Low-, or Intermediate-risk myelodysplastic syndromes in erythropoiesis stimulating agent-naive patients who require red blood cell transfusions Matteo Della Porta,1,2 Uwe Platzbecker,3 Valeria Santini,4 Guillermo Garcia-Manero,5 Rami S. Komrokji,6 Rodrigo Ito,7 Pierre Fenaux8 1Cancer Center IRCCS Humanitas Research Hospital, Milan, Italy; 2Department of Biomedical Sciences, Humanitas University, Milan, Italy; 3Medical Clinic and Policlinic 1, Hematology and Cellular Therapy, University Hospital Leipzig, Leipzig, Germany; 4Azienda Ospedaliero-Universitaria Careggi, University of Florence, Florence, Italy; 5Department of Leukemia, University of Texas MD Anderson Cancer Center, Houston, TX; 6Moffitt Cancer Center, Tampa, FL; 7Bristol Myers Squibb, Princeton, NJ; 8Service d'Hématologie Séniors, Hôpital Saint-Louis, Université Paris 7, Paris, France Presentation 2198 Presenting author disclosures M.D.P.: no conflicts of interest to disclose. 2 Introduction and objectives Introduction • Studies of epoetin alfa and darbepoetin alfa have demonstrated efficacy among patients with LR-MDS, but the patient population in which a clinically significant effect is observed may be limited1,2 • Luspatercept, a first-in-class erythroid maturation agent with a mechanism of action distinct from ESAs,3 is approved by the US FDA for the treatment of anemia failing an -

Therapeutic Class Overview Erythropoiesis-Stimulating Agents
Therapeutic Class Overview Erythropoiesis-Stimulating Agents Therapeutic Class · Overview/Summary: The erythropoiesis-stimulating agents (ESAs) currently available are darbepoetin alfa (Aranesp®), epoetin alfa (Epogen®, Procrit®) and peginesatide (Omontys®).1-4 There is no generic ESA is currently available in the United States. All ESAs are approved by the Food and Drug Administration (FDA) for the treatment of anemia in patients with chronic kidney disease; however, peginesatide is only approved for use in patients on dialysis. Darbepoetin alfa and epoetin alfa are also indicated for the treatment of anemia in cancer patients receiving chemotherapy. Moreover, epoetin alfa products are also FDA-approved for the treatment of anemia due to zidovudine therapy in patients with human immunodeficiency virus infection as well as to reduce the need for allogeneic blood transfusion in anemic patients who are at high risk for perioperative blood loss from elective, noncardiac, nonvascular surgery.1-5 Both epoetin alfa and darbepoetin alfa are manufactured via recombinant deoxyribonucleic acid technology and have similar biological effects as endogenous erythropoietin.6 Although darbepoetin alfa has identical pharmacological actions, the additional carbohydrate chains prolong its elimination half-life by two- to three-fold compared to epoetin alfa. Because of this, darbepoetin alfa is dosed less frequently compared to epoetin alfa. Peginesatide is structurally distinct from epoetin alfa and darbepoetin alfa in that it is a synthetic dimeric peptide -

Conversion Dosing Guide: from Epoetin Alfa to Aranesp® in Patients with Anemia Due to CKD on Dialysis
Conversion Dosing Guide: From epoetin alfa to Aranesp® in patients with anemia due to CKD on dialysis Indication Aranesp® (darbepoetin alfa) is indicated for the treatment of anemia due to chronic kidney disease (CKD), including patients on dialysis and patients not on dialysis. Limitations of Use • Aranesp® has not been shown to improve quality of life, fatigue, or patient well-being. • Aranesp® is not indicated for use as a substitute for red blood cell transfusions in patients who require immediate correction of anemia. Please see Important Safety Information, including Boxed WARNINGS about INCREASED RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE, on pages 2 and 3. Important Safety Information including Boxed WARNINGS • Aranesp® is contraindicated in patients with: WARNING: ESAs INCREASE THE RISK OF DEATH, – Uncontrolled hypertension MYOCARDIAL INFARCTION, STROKE, VENOUS – Pure red cell aplasia (PRCA) that begins after treatment with Aranesp® or other THROMBOEMBOLISM, THROMBOSIS OF VASCULAR erythropoietin protein drugs ACCESS AND TUMOR PROGRESSION OR RECURRENCE – Serious allergic reactions to Aranesp® Chronic Kidney Disease: • Use caution in patients with coexistent cardiovascular disease and stroke. • Patients with CKD and an insufficient hemoglobin response to ESA therapy may be • In controlled trials, patients experienced greater risks for death, serious adverse cardiovascular reactions, and at even greater risk for cardiovascular reactions and mortality than other patients. stroke when administered erythropoiesis-stimulating agents A rate of hemoglobin rise of > 1 g/dL over 2 weeks may contribute to these risks. (ESAs) to target a hemoglobin level of greater than 11 g/dL. • In controlled clinical trials, ESAs increased the risk of death in patients undergoing ® coronary artery bypass graft surgery (CABG) and the risk of deep venous • No trial has identified a hemoglobin target level, Aranesp thrombosis (DVT) in patients undergoing orthopedic procedures. -

Epoetin Alfa-Epbx
HIGHLIGHTS OF PRESCRIBING INFORMATION maintenance dose. Intravenous route recommended for patients on These highlights do not include all the information needed to use hemodialysis (2.2). RETACRIT safely and effectively. See full prescribing information for • Patients on Zidovudine due to HIV-infection: 100 Units/kg 3 times weekly RETACRIT. (2.3). RETACRIT™ (epoetin alfa-epbx) injection, for intravenous or • Patients with Cancer on Chemotherapy: 40,000 Units weekly or 150 subcutaneous use Units/kg 3 times weekly (adults); 600 Units/kg intravenously weekly (pediatric patients > 5 years) (2.4). Initial U.S. Approval: 2018 • Surgery Patients: 300 Units/kg per day daily for 15 days or 600 Units/kg RETACRIT (epoetin alfa-epbx) is biosimilar* to EPOGEN/PROCRIT weekly (2.5). (epoetin alfa) WARNING: ESAs INCREASE THE RISK OF DEATH, -------------------- DOSAGE FORMS AND STRENGTHS -------------------- MYOCARDIAL INFARCTION, STROKE, VENOUS Injection THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS 2,000 Units/mL, 3,000 Units/mL, 4,000 Units/mL, 10,000 Units/mL, and AND TUMOR PROGRESSION OR RECURRENCE 40,000 Units/mL in single-dose vials (3) See full prescribing information for complete boxed warning. ------------------------------CONTRAINDICATIONS----------------------------- Chronic Kidney Disease: • Uncontrolled hypertension (4) • In controlled trials, patients experienced greater risks for death, • Pure red cell aplasia (PRCA) that begins after treatment with RETACRIT serious adverse cardiovascular reactions, and stroke when or other erythropoietin protein drugs (4) administered erythropoiesis-stimulating agents (ESAs) to target a • Serious allergic reactions to RETACRIT or other epoetin alfa products (4) hemoglobin level of greater than 11 g/dL (5.1). • No trial has identified a hemoglobin target level, ESA dose, or dosing -----------------------WARNINGS AND PRECAUTIONS----------------------- strategy that does not increase these risks. -

Aranesp, INN-Darbepoetin Alfa
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Aranesp 10 micrograms solution for injection in pre-filled syringe. Aranesp 15 micrograms solution for injection in pre-filled syringe. Aranesp 20 micrograms solution for injection in pre-filled syringe. Aranesp 30 micrograms solution for injection in pre-filled syringe. Aranesp 40 micrograms solution for injection in pre-filled syringe. Aranesp 50 micrograms solution for injection in pre-filled syringe. Aranesp 60 micrograms solution for injection in pre-filled syringe. Aranesp 80 micrograms solution for injection in pre-filled syringe. Aranesp 100 micrograms solution for injection in pre-filled syringe. Aranesp 130 micrograms solution for injection in pre-filled syringe. Aranesp 150 micrograms solution for injection in pre-filled syringe. Aranesp 300 micrograms solution for injection in pre-filled syringe. Aranesp 500 micrograms solution for injection in pre-filled syringe. Aranesp 10 micrograms solution for injection in pre-filled pen. Aranesp 15 micrograms solution for injection in pre-filled pen. Aranesp 20 micrograms solution for injection in pre-filled pen. Aranesp 30 micrograms solution for injection in pre-filled pen. Aranesp 40 micrograms solution for injection in pre-filled pen. Aranesp 50 micrograms solution for injection in pre-filled pen. Aranesp 60 micrograms solution for injection in pre-filled pen. Aranesp 80 micrograms solution for injection in pre-filled pen. Aranesp 100 micrograms solution for injection in pre-filled pen. Aranesp 130 micrograms solution for injection in pre-filled pen. Aranesp 150 micrograms solution for injection in pre-filled pen. Aranesp 300 micrograms solution for injection in pre-filled pen. -

Coding Medical Necessity: Erythropoiesis Stimulating Agents (Esas)
Coding Medical Necessity: Erythropoiesis Stimulating Agents (ESAs) This article contains instructions for coding medical necessity in accordance with both the national coverage determination (NCD) and local coverage determination (LCD) and other CMS instructions on darbepoetin alfa (Aranesp®, DPA) and epoetin alfa (Epogen®, Procrit®, EPO). These coding guidelines are not intended to replace any found in the ICD-9-CM Official Guidelines for Coding and Reporting, nor are they intended to provide guidance on when a condition should be coded. Rather, this article should be used in conjunction with the UB-04 Data Specifications Manual and the ICD-9-CM Official Guidelines for Coding and Reporting. This article supersedes all previous articles on this subject. Providers should refer to CMS manuals and updates issued in Change Requests for additional claim form-specific billing instructions, including, but not limited to modifiers, necessary for payment. General Information for all claims for ESAs: These coding guidelines specifically address the documentation of medical necessity on the claim, i.e., the coding in this guidance must be used to indicate the conditions that convey medical necessity of the drug treatment. Providers may not code a claim with more than one drug code (J or Q) for DPA or EPO, i.e., only one of the DPA or EPO codes may appear on a claim. The administration of this class of drugs should NOT be billed using any of the chemotherapy administration codes. Providers should use the appropriate therapeutic, prophylactic, and diagnostic injections and infusions code. No payment can be made for drugs when self-administered or administered by a caregiver (except for drugs administered under the auspices of the ESRD program). -

Aranesp, INN-Darbepoetin Alfa
SCIENTIFIC DISCUSSION This module reflects the initial scientific discussion for the approval of Aranesp. This scientific discussion has been updated until 1 September 2003. For scientific information on procedures after this date please refer to module 8B. 1. Introduction Patients with chronic renal failure (CRF) develop uremic anaemia as one of the most obvious signs of the disease. This symptom is caused by impeded renal production of erythropoietin (EPO). EPO controls red blood cell counts (RBC) production by promoting survival, proliferation and differentiation of erythroid progenitors in the bone marrow. Effective management of anaemia in chronic renal failure (CRF) has a major impact on quality of life and may influence survival. Supplementation with recombinant human erythropoietin (r-HuEPO) is currently the standard treatment for anaemia in those patients. It is regarded as an effective drug, which has an established safety record. Recombinant human erythropoietin is currently available as a treatment for anaemia in end stage renal disease. Administration 2 to 3 times weekly is required in the majority of subjects. The aim of inventing this new molecular entity of darbepoetin alfa was to obtain a therapeutic with a longer biological half-life compared to r-HuEPO, allowing a reduction of the frequency of injections necessary to maintain a desired level of systemic haemoglobin and haematocrit. The chronic nature of CRF (unless a subject receives a kidney transplant) means that treatment may continue for a long part of the subject’s life and multiple weekly injections of r-HuEPO can have a major impact on subjects and care givers. Research has indicated that the sialic acid containing carbohydrate of erythropoietin determines its serum half-live. -

Aranesp® (Darbepoetin Alfa)
Aranesp® (darbepoetin alfa) Aranesp (darbepoetin alfa) is a man-made protein that helps your body produce red blood cells (RBC’s). This protein may be reduced because of kidney failure or with use of certain medications. When fewer red blood cells are produced, you can develop a condition called anemia. Aranesp is used to treat anemia caused by chemotherapy or chronic kidney disease. How do I take Aranesp? • Aranesp is injected under the skin, it is given most often in clinic by a nurse or doctor. • It is given every 1 to 4 weeks depending on the condition being treated and as prescribed by your doctor. • You may need frequent medical tests to be sure that there are no harmful effects from the use of Aranesp. Your injections may be delayed based on the results of these tests. • If you need to have surgery, make your surgeon aware that you are using Aranesp. You may need to stop using for a period of time. What drugs interact with Aranesp? Other drugs may interact with Aranesp including: • Prescription and over-the-counter medicines • Vitamins • Herbal products Tell each of your health care providers about all medicines you use now and any medicine you start or stop using. Department of Nephrology - 1 - What should I discuss with my healthcare provider before using Aranesp? 1. You should not use this medication is you are allergic to Aranesp; or if allergic to Epogen or Procrit (epoetin alfa) 2. To make sure that Aranesp is safe for you, tell your healthcare provider if you have: • Heart disease, congestive heart failure, high blood pressure -

Amgen 2010 Annual Report and Financial Summary
Amgen 2010 Annual Report and Financial Summary 3/9/11 12:32 PM Amgen Mission To serve patients About Amgen Amgen Values Amgen discovers, develops, manufactures, and delivers innovative human therapeutics. A biotechnology pioneer since 1980, Amgen was Be science-based one of the fi rst companies to realize the new science’s promise by Compete intensely and win bringing safe, effective medicines from lab to manufacturing plant to Create value for patients, staff, and stockholders patient. Amgen therapeutics have changed the practice of medicine, Be ethical helping millions of people around the world in the fi ght against cancer, Trust and respect each other kidney disease, rheumatoid arthritis, bone disease, and other serious Ensure quality illnesses. With a deep and broad pipeline of potential new medicines, Work in teams Amgen remains committed to advancing science to dramatically Collaborate, communicate, improve people’s lives. and be accountable 28822_Cov.indd 2 Letter to Stockholders Robert A. Bradway, Kevin W. Sharer, President and Chairman and Chief Operating Offi cer Chief Executive Offi cer Dear Stockholders, 2010 was a year of challenges met and promises delivered for Amgen’s Products patients, stockholders, and staff. We delivered an important, innovative bone health therapy, denosumab, in the form of two novel medicines: Prolia®, Aranesp® (darbepoetin alfa) approved in Europe, the United States, and several other countries for ™ postmenopausal osteoporosis, and XGEVA , approved in the United States Enbrel® (etanercept) for the prevention of skeletal-related events in patients with bone metastases from solid tumors. We grew revenues 3 percent, adjusted earnings per EPOGEN® (Epoetin alfa) share* 6 percent, and generated nearly $6 billion in operating cash fl ow, ® despite continuing global economic turmoil and a $200 million adverse Neulasta (pegfi lgrastim) impact to revenues from U.S.