Human Protein Product Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer
ORIGINAL RESEARCH published: 15 April 2021 doi: 10.3389/fonc.2021.566539 Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer Qinglian He 1, Ziqi Li 1, Jinbao Yin 1, Yuling Li 2, Yuting Yin 1, Xue Lei 1 and Wei Zhu 1* 1 Department of Pathology, Guangdong Medical University, Dongguan, China, 2 Department of Pathology, Dongguan People’s Hospital, Southern Medical University, Dongguan, China Background: Colorectal cancer (CRC) is a common malignant solid tumor with an extremely low survival rate after relapse. Previous investigations have shown that autophagy possesses a crucial function in tumors. However, there is no consensus on the value of autophagy-associated genes in predicting the prognosis of CRC patients. Edited by: This work screens autophagy-related markers and signaling pathways that may Fenglin Liu, Fudan University, China participate in the development of CRC, and establishes a prognostic model of CRC Reviewed by: based on autophagy-associated genes. Brian M. Olson, Emory University, United States Methods: Gene transcripts from the TCGA database and autophagy-associated gene Zhengzhi Zou, data from the GeneCards database were used to obtain expression levels of autophagy- South China Normal University, China associated genes, followed by Wilcox tests to screen for autophagy-related differentially Faqing Tian, Longgang District People's expressed genes. Then, 11 key autophagy-associated genes were identified through Hospital of Shenzhen, China univariate and multivariate Cox proportional hazard regression analysis and used to Yibing Chen, Zhengzhou University, China establish prognostic models. Additionally, immunohistochemical and CRC cell line data Jian Tu, were used to evaluate the results of our three autophagy-associated genes EPHB2, University of South China, China NOL3, and SNAI1 in TCGA. -

Familial Cortical Myoclonus Caused by Mutation in NOL3 by Jonathan Foster Rnsseil DISSERTATION Submitted in Partial Satisfaction
Familial Cortical Myoclonus Caused by Mutation in NOL3 by Jonathan Foster Rnsseil DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences in the Copyright 2013 by Jonathan Foster Russell ii I dedicate this dissertation to Mom and Dad, for their adamantine love and support iii No man has earned the right to intellectual ambition until he has learned to lay his course by a star which he has never seen—to dig by the divining rod for springs which he may never reach. In saying this, I point to that which will make your study heroic. For I say to you in all sadness of conviction, that to think great thoughts you must be heroes as well as idealists. Only when you have worked alone – when you have felt around you a black gulf of solitude more isolating than that which surrounds the dying man, and in hope and in despair have trusted to your own unshaken will – then only will you have achieved. Thus only can you gain the secret isolated joy of the thinker, who knows that, a hundred years after he is dead and forgotten, men who never heard of him will be moving to the measure of his thought—the subtile rapture of a postponed power, which the world knows not because it has no external trappings, but which to his prophetic vision is more real than that which commands an army. -Oliver Wendell Holmes, Jr. iv ACKNOWLEDGMENTS I am humbled by the efforts of many, many others who were essential for this work. -
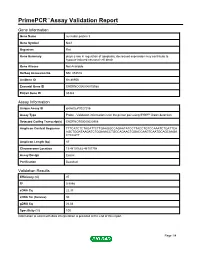
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nucleolar protein 3 Gene Symbol Nol3 Organism Rat Gene Summary plays a role in regulation of apoptosis; decreased expression may contribute to hypoxia-induced neuronal cell death Gene Aliases Not Available RefSeq Accession No. NM_053516 UniGene ID Rn.86956 Ensembl Gene ID ENSRNOG00000015588 Entrez Gene ID 85383 Assay Information Unique Assay ID qRnoCEP0027256 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000020908 Amplicon Context Sequence TTTCATCTCTAGATTCTTGAAGGCCAGAATATCCTTACCTGTCCAAATCTGATTCA AGCTGGATAAGATCTGGAAACCTGCCAGAACTGGACCAAGTCAATGCAGCAAGA CTCCATT Amplicon Length (bp) 87 Chromosome Location 19:48101682-48101798 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9998 cDNA Cq 22.33 cDNA Tm (Celsius) 80 gDNA Cq 26.04 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Nol3, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Medical Relevance of Protein-Truncating Variants Across 337,205 Individuals in the UK Biobank Study
ARTICLE DOI: 10.1038/s41467-018-03910-9 OPEN Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study Christopher DeBoever1,2, Yosuke Tanigawa 1, Malene E. Lindholm3, Greg McInnes1, Adam Lavertu1, Erik Ingelsson4, Chris Chang3, Euan A. Ashley 5, Carlos D. Bustamante1,2, Mark J. Daly 6,7 & Manuel A. Rivas1 Protein-truncating variants can have profound effects on gene function and are critical for 1234567890():,; clinical genome interpretation and generating therapeutic hypotheses, but their relevance to medical phenotypes has not been systematically assessed. Here, we characterize the effect of 18,228 protein-truncating variants across 135 phenotypes from the UK Biobank and find 27 associations between medical phenotypes and protein-truncating variants in genes outside the major histocompatibility complex. We perform phenome-wide analyses and directly measure the effect in homozygous carriers, commonly referred to as “human knockouts,” across medical phenotypes for genes implicated as being protective against disease or associated with at least one phenotype in our study. We find several genes with strong pleiotropic or non-additive effects. Our results illustrate the importance of protein-truncating variants in a variety of diseases. 1 Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University, Stanford, CA 94305, USA. 3 Grail, Inc., 1525 O’Brien Drive, Menlo Park, CA 94025, USA. 4 Division of Cardiovascular Medicine, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA. 5 Department of Medicine, School of Medicine, Stanford University, Stanford, CA 94305, USA. 6 Analytical and Translational Genetics Unit, Boston, MA 02114, USA. -

Wppi: Weighting Protein-Protein Interactions
Package ‘wppi’ September 23, 2021 Type Package Title Weighting protein-protein interactions Version 1.1.2 Description Protein-protein interaction data is essential for omics data analysis and modeling. Database knowledge is general, not specific for cell type, physiological condition or any other context determining which connections are functional and contribute to the signaling. Functional annotations such as Gene Ontology and Human Phenotype Ontology might help to evaluate the relevance of interactions. This package predicts functional relevance of protein-protein interactions based on functional annotations such as Human Protein Ontology and Gene Ontology, and prioritizes genes based on network topology, functional scores and a path search algorithm. License MIT + file LICENSE URL https://github.com/AnaGalhoz37/wppi BugReports https://github.com/AnaGalhoz37/wppi/issues biocViews GraphAndNetwork, Network, Pathways, Software, GeneSignaling, GeneTarget, SystemsBiology, Transcriptomics, Annotation Encoding UTF-8 VignetteBuilder knitr Depends R(>= 4.1) Imports dplyr, igraph, logger, methods, magrittr, Matrix, OmnipathR(>= 2.99.8), progress, purrr, rlang, RCurl, stats, tibble, tidyr Suggests knitr, testthat, rmarkdown RoxygenNote 7.1.1 NeedsCompilation no git_url https://git.bioconductor.org/packages/wppi git_branch master git_last_commit 6aa91be git_last_commit_date 2021-07-15 1 2 common_neighbors Date/Publication 2021-09-23 Author Ana Galhoz [cre, aut] (<https://orcid.org/0000-0001-7402-5292>), Denes Turei [aut] (<https://orcid.org/0000-0002-7249-9379>), Michael P. Menden [aut] (<https://orcid.org/0000-0003-0267-5792>), Albert Krewinkel [ctb, cph] (pagebreak Lua filter) Maintainer Ana Galhoz <[email protected]> R topics documented: common_neighbors . .2 count_genes . .3 filter_annot_with_network . .4 functional_annot . .5 graph_from_op . .6 in_omnipath . .6 prioritization_genes . .7 process_annot . -

Molecular Cytogenetic Characterization of Partial Monosomy 2P and Trisomy 16Q in a Newborn: a Case Report
EXPERIMENTAL AND THERAPEUTIC MEDICINE 18: 1267-1275, 2019 Molecular cytogenetic characterization of partial monosomy 2p and trisomy 16q in a newborn: A case report FAGUI YUE1,2, YUTING JIANG1,2, YUAN PAN1,2, LEILEI LI1,2, LINLIN LI1,2, RUIZHI LIU1,2 and RUIXUE WANG1,2 1Center for Reproductive Medicine and Center for Prenatal Diagnosis, The First Hospital; 2Jilin Engineering Research Center for Reproductive Medicine and Genetics, Jilin University, Changchun, Jilin 130021, P.R. China Received August 12, 2018; Accepted May 16, 2019 DOI: 10.3892/etm.2019.7695 Abstract. Trisomy 16q is a rare disorder with severe abnor- Introduction malities, which always leads to early postnatal mortality. It usually results from a parental translocation, exhibiting 16q Trisomy 16 is recognized as the most common type of trisomy duplication associated with another chromosomal deletion. in first‑trimester spontaneous abortions, occurring in 1% of The present study reports on the clinical presentation and all clinically recognized pregnancies, while being rarer in the molecular cytogenetic results of a small-for-gestational-age second and third������������������������������������������������ trimesters����������������������������������������������� (1-3). Early lethality and incompat- infant, consisting of partial trisomy 16q21→qter and mono- ibility with life have been described as its major outcomes (1). somy 2p25.3→pter. The proband presented with moderately Trisomy 16 may be classified into three major types: Full low birthweight, small anterior fontanelles, prominent trisomy, mosaics and partial trisomy of 16p or 16q. Since forehead, low hairline, telecanthus, flat nasal bridge, choanal Schmickel (����)������ ��re�orted the fifirst rst case of ���16q������������������� trisomy as identi- atresia, clinodactyly of the fifth fingers, urogenital anomalies, fied using a chromosome banding techni�ue in �975, >30 cases congenital muscular torticollis and congenital laryngoma- of partial trisomy 16q have been described. -

A Graph-Theoretic Approach to Model Genomic Data and Identify Biological Modules Asscociated with Cancer Outcomes
A Graph-Theoretic Approach to Model Genomic Data and Identify Biological Modules Asscociated with Cancer Outcomes Deanna Petrochilos A dissertation presented in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2013 Reading Committee: Neil Abernethy, Chair John Gennari, Ali Shojaie Program Authorized to Offer Degree: Biomedical Informatics and Health Education UMI Number: 3588836 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMI 3588836 Published by ProQuest LLC (2013). Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, MI 48106 - 1346 ©Copyright 2013 Deanna Petrochilos University of Washington Abstract Using Graph-Based Methods to Integrate and Analyze Cancer Genomic Data Deanna Petrochilos Chair of the Supervisory Committee: Assistant Professor Neil Abernethy Biomedical Informatics and Health Education Studies of the genetic basis of complex disease present statistical and methodological challenges in the discovery of reliable and high-confidence genes that reveal biological phenomena underlying the etiology of disease or gene signatures prognostic of disease outcomes. This dissertation examines the capacity of graph-theoretical methods to model and analyze genomic information and thus facilitate using prior knowledge to create a more discrete and functionally relevant feature space. -

Selecting the Most Appropriate Time Points to Profile in High-Throughput Studies
Selecting the most appropriate time points to profile in high-throughput studies Michael Kleyman*1, Emre Sefer*1, Teodora Nicola2, Celia Espinoza3, Divya Chhabra3, James S. Hagood3, Naftali Kaminski4, Namasivayam Ambalavanan2, Ziv-Bar Joseph1 *Equal contribution 1 Machine Learning and Computational Biology, School of Computer Science, Carnegie Mellon Univer- sity, Pittsburgh, PA, USA 2 Division of Neonatology, Dept of Pediatrics, University of Alabama at Birmingham, Birmingham, AL, USA 3 Division of Respiratory Medicine, Dept of Pediatrics, University of California San Diego, La Jolla, CARady Children's Hospital San Diego, San Diego, CA 4 Section of Pulmonary, Critical Care and Sleep Medicine, School of Medicine, Yale University, New Heaven, CT E-mail: [email protected] Abstract Biological systems are increasingly being studied by high throughput profiling of molecular data over time. Determining the set of time points to sample in studies that profile several different types of molecular data is still challenging. Here we present the Time Point Selection (TPS) method that solves this combinatorial problem in a principled and practical way. TPS utilizes expression data from a small set of genes sampled at a high rate. As we show by applying TPS to study mouse lung development, the points selected by TPS can be used to reconstruct an accurate representation for the expression values of the non selected points. Further, even though the selection is only based on gene expression, these points are also appropriate for representing a much larger set of protein, miRNA and DNA methylation changes over time. TPS can thus serve as a key design strategy for high throughput time series experiments. -

Alterations of the Pro-Survival Bcl-2 Protein Interactome in Breast Cancer
bioRxiv preprint doi: https://doi.org/10.1101/695379; this version posted July 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Alterations of the pro-survival Bcl-2 protein interactome in 2 breast cancer at the transcriptional, mutational and 3 structural level 4 5 Simon Mathis Kønig1, Vendela Rissler1, Thilde Terkelsen1, Matteo Lambrughi1, Elena 6 Papaleo1,2 * 7 1Computational Biology Laboratory, Danish Cancer Society Research Center, 8 Strandboulevarden 49, 2100, Copenhagen 9 10 2Translational Disease Systems Biology, Faculty of Health and Medical Sciences, Novo 11 Nordisk Foundation Center for Protein Research University of Copenhagen, Copenhagen, 12 Denmark 13 14 Abstract 15 16 Apoptosis is an essential defensive mechanism against tumorigenesis. Proteins of the B-cell 17 lymphoma-2 (Bcl-2) family regulates programmed cell death by the mitochondrial apoptosis 18 pathway. In response to intracellular stresses, the apoptotic balance is governed by interactions 19 of three distinct subgroups of proteins; the activator/sensitizer BH3 (Bcl-2 homology 3)-only 20 proteins, the pro-survival, and the pro-apoptotic executioner proteins. Changes in expression 21 levels, stability, and functional impairment of pro-survival proteins can lead to an imbalance 22 in tissue homeostasis. Their overexpression or hyperactivation can result in oncogenic effects. 23 Pro-survival Bcl-2 family members carry out their function by binding the BH3 short linear 24 motif of pro-apoptotic proteins in a modular way, creating a complex network of protein- 25 protein interactions. -

And Post-Translationally N-Myristoylated Proteomes in Human Cells
ARTICLE Received 5 Mar 2014 | Accepted 5 Aug 2014 | Published 26 Sep 2014 DOI: 10.1038/ncomms5919 OPEN Global profiling of co- and post-translationally N-myristoylated proteomes in human cells Emmanuelle Thinon1,2,*,w, Remigiusz A. Serwa1,*, Malgorzata Broncel1, James A. Brannigan3, Ute Brassat1,2,w, Megan H. Wright1,4,w, William P. Heal1,w, Anthony J. Wilkinson3, David J. Mann2,4 & Edward W. Tate1,4 Protein N-myristoylation is a ubiquitous co- and post-translational modification that has been implicated in the development and progression of a range of human diseases. Here, we report the global N-myristoylated proteome in human cells determined using quantitative chemical proteomics combined with potent and specific human N-myristoyltransferase (NMT) inhibition. Global quantification of N-myristoylation during normal growth or apoptosis allowed the identification of 4100 N-myristoylated proteins, 495% of which are identified for the first time at endogenous levels. Furthermore, quantitative dose response for inhibition of N-myristoylation is determined for 470 substrates simultaneously across the proteome. Small-molecule inhibition through a conserved substrate-binding pocket is also demonstrated by solving the crystal structures of inhibitor-bound NMT1 and NMT2. The presented data substantially expand the known repertoire of co- and post-translational N-myristoylation in addition to validating tools for the pharmacological inhibition of NMT in living cells. 1 Department of Chemistry, Imperial College London, Exhibition Road, London SW7 2AZ, UK. 2 Department of Life Sciences, Imperial College London, Exhibition Road, London SW7 2AZ, UK. 3 York Structural Biology Laboratory, Department of Chemistry, University of York, York YO10 5DD, UK. -

393LN V 393P 344SQ V 393P Probe Set Entrez Gene
393LN v 393P 344SQ v 393P Entrez fold fold probe set Gene Gene Symbol Gene cluster Gene Title p-value change p-value change chemokine (C-C motif) ligand 21b /// chemokine (C-C motif) ligand 21a /// chemokine (C-C motif) ligand 21c 1419426_s_at 18829 /// Ccl21b /// Ccl2 1 - up 393 LN only (leucine) 0.0047 9.199837 0.45212 6.847887 nuclear factor of activated T-cells, cytoplasmic, calcineurin- 1447085_s_at 18018 Nfatc1 1 - up 393 LN only dependent 1 0.009048 12.065 0.13718 4.81 RIKEN cDNA 1453647_at 78668 9530059J11Rik1 - up 393 LN only 9530059J11 gene 0.002208 5.482897 0.27642 3.45171 transient receptor potential cation channel, subfamily 1457164_at 277328 Trpa1 1 - up 393 LN only A, member 1 0.000111 9.180344 0.01771 3.048114 regulating synaptic membrane 1422809_at 116838 Rims2 1 - up 393 LN only exocytosis 2 0.001891 8.560424 0.13159 2.980501 glial cell line derived neurotrophic factor family receptor alpha 1433716_x_at 14586 Gfra2 1 - up 393 LN only 2 0.006868 30.88736 0.01066 2.811211 1446936_at --- --- 1 - up 393 LN only --- 0.007695 6.373955 0.11733 2.480287 zinc finger protein 1438742_at 320683 Zfp629 1 - up 393 LN only 629 0.002644 5.231855 0.38124 2.377016 phospholipase A2, 1426019_at 18786 Plaa 1 - up 393 LN only activating protein 0.008657 6.2364 0.12336 2.262117 1445314_at 14009 Etv1 1 - up 393 LN only ets variant gene 1 0.007224 3.643646 0.36434 2.01989 ciliary rootlet coiled- 1427338_at 230872 Crocc 1 - up 393 LN only coil, rootletin 0.002482 7.783242 0.49977 1.794171 expressed sequence 1436585_at 99463 BB182297 1 - up 393