Demography-Adjusted Tests of Neutrality Based on Genome-Wide SNP Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Epigenetic Silencing of TGFBI Confers Resistance to Trastuzumab in Human Breast Cancer
Palomeras et al. Breast Cancer Research (2019) 21:79 https://doi.org/10.1186/s13058-019-1160-x RESEARCH ARTICLE Open Access Epigenetic silencing of TGFBI confers resistance to trastuzumab in human breast cancer Sònia Palomeras1†, Ángel Diaz-Lagares2,3†, Gemma Viñas1,4,5, Fernando Setien2, Humberto J. Ferreira2, Glòria Oliveras1,6, Ana B. Crujeiras7,8, Alejandro Hernández4, David H. Lum9, Alana L. Welm9, Manel Esteller2,10,11,12,13* and Teresa Puig1* Abstract Background: Acquired resistance to trastuzumab is a major clinical problem in the treatment of HER2-positive (HER2+) breast cancer patients. The selection of trastuzumab-resistant patients is a great challenge of precision oncology. The aim of this study was to identify novel epigenetic biomarkers associated to trastuzumab resistance in HER2+ BC patients. Methods: We performed a genome-wide DNA methylation (450K array) and a transcriptomic analysis (RNA-Seq) comparing trastuzumab-sensitive (SK) and trastuzumab-resistant (SKTR) HER2+ human breast cancer cell models. The methylation and expression levels of candidate genes were validated by bisulfite pyrosequencing and qRT-PCR, respectively. Functional assays were conducted in the SK and SKTR models by gene silencing and overexpression. Methylation analysis in 24 HER2+ human BC samples with complete response or non-response to trastuzumab- based treatment was conducted by bisulfite pyrosequencing. Results: Epigenomic and transcriptomic analysis revealed the consistent hypermethylation and downregulation of TGFBI, CXCL2, and SLC38A1 genes in association with trastuzumab resistance. The DNA methylation and expression levels of these genes were validated in both sensitive and resistant models analyzed. Of the genes, TGFBI presented the highest hypermethylation-associated silencing both at the transcriptional and protein level. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer
ORIGINAL RESEARCH published: 15 April 2021 doi: 10.3389/fonc.2021.566539 Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer Qinglian He 1, Ziqi Li 1, Jinbao Yin 1, Yuling Li 2, Yuting Yin 1, Xue Lei 1 and Wei Zhu 1* 1 Department of Pathology, Guangdong Medical University, Dongguan, China, 2 Department of Pathology, Dongguan People’s Hospital, Southern Medical University, Dongguan, China Background: Colorectal cancer (CRC) is a common malignant solid tumor with an extremely low survival rate after relapse. Previous investigations have shown that autophagy possesses a crucial function in tumors. However, there is no consensus on the value of autophagy-associated genes in predicting the prognosis of CRC patients. Edited by: This work screens autophagy-related markers and signaling pathways that may Fenglin Liu, Fudan University, China participate in the development of CRC, and establishes a prognostic model of CRC Reviewed by: based on autophagy-associated genes. Brian M. Olson, Emory University, United States Methods: Gene transcripts from the TCGA database and autophagy-associated gene Zhengzhi Zou, data from the GeneCards database were used to obtain expression levels of autophagy- South China Normal University, China associated genes, followed by Wilcox tests to screen for autophagy-related differentially Faqing Tian, Longgang District People's expressed genes. Then, 11 key autophagy-associated genes were identified through Hospital of Shenzhen, China univariate and multivariate Cox proportional hazard regression analysis and used to Yibing Chen, Zhengzhou University, China establish prognostic models. Additionally, immunohistochemical and CRC cell line data Jian Tu, were used to evaluate the results of our three autophagy-associated genes EPHB2, University of South China, China NOL3, and SNAI1 in TCGA. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Familial Cortical Myoclonus Caused by Mutation in NOL3 by Jonathan Foster Rnsseil DISSERTATION Submitted in Partial Satisfaction
Familial Cortical Myoclonus Caused by Mutation in NOL3 by Jonathan Foster Rnsseil DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences in the Copyright 2013 by Jonathan Foster Russell ii I dedicate this dissertation to Mom and Dad, for their adamantine love and support iii No man has earned the right to intellectual ambition until he has learned to lay his course by a star which he has never seen—to dig by the divining rod for springs which he may never reach. In saying this, I point to that which will make your study heroic. For I say to you in all sadness of conviction, that to think great thoughts you must be heroes as well as idealists. Only when you have worked alone – when you have felt around you a black gulf of solitude more isolating than that which surrounds the dying man, and in hope and in despair have trusted to your own unshaken will – then only will you have achieved. Thus only can you gain the secret isolated joy of the thinker, who knows that, a hundred years after he is dead and forgotten, men who never heard of him will be moving to the measure of his thought—the subtile rapture of a postponed power, which the world knows not because it has no external trappings, but which to his prophetic vision is more real than that which commands an army. -Oliver Wendell Holmes, Jr. iv ACKNOWLEDGMENTS I am humbled by the efforts of many, many others who were essential for this work. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -
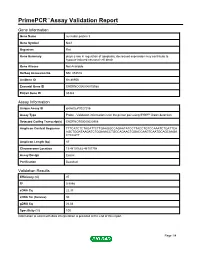
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nucleolar protein 3 Gene Symbol Nol3 Organism Rat Gene Summary plays a role in regulation of apoptosis; decreased expression may contribute to hypoxia-induced neuronal cell death Gene Aliases Not Available RefSeq Accession No. NM_053516 UniGene ID Rn.86956 Ensembl Gene ID ENSRNOG00000015588 Entrez Gene ID 85383 Assay Information Unique Assay ID qRnoCEP0027256 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000020908 Amplicon Context Sequence TTTCATCTCTAGATTCTTGAAGGCCAGAATATCCTTACCTGTCCAAATCTGATTCA AGCTGGATAAGATCTGGAAACCTGCCAGAACTGGACCAAGTCAATGCAGCAAGA CTCCATT Amplicon Length (bp) 87 Chromosome Location 19:48101682-48101798 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9998 cDNA Cq 22.33 cDNA Tm (Celsius) 80 gDNA Cq 26.04 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Nol3, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Table SII. Significantly Differentially Expressed Mrnas of GSE23558 Data Series with the Criteria of Adjusted P<0.05 And
Table SII. Significantly differentially expressed mRNAs of GSE23558 data series with the criteria of adjusted P<0.05 and logFC>1.5. Probe ID Adjusted P-value logFC Gene symbol Gene title A_23_P157793 1.52x10-5 6.91 CA9 carbonic anhydrase 9 A_23_P161698 1.14x10-4 5.86 MMP3 matrix metallopeptidase 3 A_23_P25150 1.49x10-9 5.67 HOXC9 homeobox C9 A_23_P13094 3.26x10-4 5.56 MMP10 matrix metallopeptidase 10 A_23_P48570 2.36x10-5 5.48 DHRS2 dehydrogenase A_23_P125278 3.03x10-3 5.40 CXCL11 C-X-C motif chemokine ligand 11 A_23_P321501 1.63x10-5 5.38 DHRS2 dehydrogenase A_23_P431388 2.27x10-6 5.33 SPOCD1 SPOC domain containing 1 A_24_P20607 5.13x10-4 5.32 CXCL11 C-X-C motif chemokine ligand 11 A_24_P11061 3.70x10-3 5.30 CSAG1 chondrosarcoma associated gene 1 A_23_P87700 1.03x10-4 5.25 MFAP5 microfibrillar associated protein 5 A_23_P150979 1.81x10-2 5.25 MUCL1 mucin like 1 A_23_P1691 2.71x10-8 5.12 MMP1 matrix metallopeptidase 1 A_23_P350005 2.53x10-4 5.12 TRIML2 tripartite motif family like 2 A_24_P303091 1.23x10-3 4.99 CXCL10 C-X-C motif chemokine ligand 10 A_24_P923612 1.60x10-5 4.95 PTHLH parathyroid hormone like hormone A_23_P7313 6.03x10-5 4.94 SPP1 secreted phosphoprotein 1 A_23_P122924 2.45x10-8 4.93 INHBA inhibin A subunit A_32_P155460 6.56x10-3 4.91 PICSAR P38 inhibited cutaneous squamous cell carcinoma associated lincRNA A_24_P686965 8.75x10-7 4.82 SH2D5 SH2 domain containing 5 A_23_P105475 7.74x10-3 4.70 SLCO1B3 solute carrier organic anion transporter family member 1B3 A_24_P85099 4.82x10-5 4.67 HMGA2 high mobility group AT-hook 2 A_24_P101651 -

Medical Relevance of Protein-Truncating Variants Across 337,205 Individuals in the UK Biobank Study
ARTICLE DOI: 10.1038/s41467-018-03910-9 OPEN Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study Christopher DeBoever1,2, Yosuke Tanigawa 1, Malene E. Lindholm3, Greg McInnes1, Adam Lavertu1, Erik Ingelsson4, Chris Chang3, Euan A. Ashley 5, Carlos D. Bustamante1,2, Mark J. Daly 6,7 & Manuel A. Rivas1 Protein-truncating variants can have profound effects on gene function and are critical for 1234567890():,; clinical genome interpretation and generating therapeutic hypotheses, but their relevance to medical phenotypes has not been systematically assessed. Here, we characterize the effect of 18,228 protein-truncating variants across 135 phenotypes from the UK Biobank and find 27 associations between medical phenotypes and protein-truncating variants in genes outside the major histocompatibility complex. We perform phenome-wide analyses and directly measure the effect in homozygous carriers, commonly referred to as “human knockouts,” across medical phenotypes for genes implicated as being protective against disease or associated with at least one phenotype in our study. We find several genes with strong pleiotropic or non-additive effects. Our results illustrate the importance of protein-truncating variants in a variety of diseases. 1 Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University, Stanford, CA 94305, USA. 3 Grail, Inc., 1525 O’Brien Drive, Menlo Park, CA 94025, USA. 4 Division of Cardiovascular Medicine, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA. 5 Department of Medicine, School of Medicine, Stanford University, Stanford, CA 94305, USA. 6 Analytical and Translational Genetics Unit, Boston, MA 02114, USA. -

Wppi: Weighting Protein-Protein Interactions
Package ‘wppi’ September 23, 2021 Type Package Title Weighting protein-protein interactions Version 1.1.2 Description Protein-protein interaction data is essential for omics data analysis and modeling. Database knowledge is general, not specific for cell type, physiological condition or any other context determining which connections are functional and contribute to the signaling. Functional annotations such as Gene Ontology and Human Phenotype Ontology might help to evaluate the relevance of interactions. This package predicts functional relevance of protein-protein interactions based on functional annotations such as Human Protein Ontology and Gene Ontology, and prioritizes genes based on network topology, functional scores and a path search algorithm. License MIT + file LICENSE URL https://github.com/AnaGalhoz37/wppi BugReports https://github.com/AnaGalhoz37/wppi/issues biocViews GraphAndNetwork, Network, Pathways, Software, GeneSignaling, GeneTarget, SystemsBiology, Transcriptomics, Annotation Encoding UTF-8 VignetteBuilder knitr Depends R(>= 4.1) Imports dplyr, igraph, logger, methods, magrittr, Matrix, OmnipathR(>= 2.99.8), progress, purrr, rlang, RCurl, stats, tibble, tidyr Suggests knitr, testthat, rmarkdown RoxygenNote 7.1.1 NeedsCompilation no git_url https://git.bioconductor.org/packages/wppi git_branch master git_last_commit 6aa91be git_last_commit_date 2021-07-15 1 2 common_neighbors Date/Publication 2021-09-23 Author Ana Galhoz [cre, aut] (<https://orcid.org/0000-0001-7402-5292>), Denes Turei [aut] (<https://orcid.org/0000-0002-7249-9379>), Michael P. Menden [aut] (<https://orcid.org/0000-0003-0267-5792>), Albert Krewinkel [ctb, cph] (pagebreak Lua filter) Maintainer Ana Galhoz <[email protected]> R topics documented: common_neighbors . .2 count_genes . .3 filter_annot_with_network . .4 functional_annot . .5 graph_from_op . .6 in_omnipath . .6 prioritization_genes . .7 process_annot . -

Micrornas Link the Germinal Center Reaction and B Cell Lymphomagenesis
Universidad Autónoma de Madrid Departamento de Bioquímica microRNAs link the Germinal Center reaction and B cell lymphomagenesis PhD Thesis Nahikari Bartolomé Izquierdo Madrid, 2016 Departamento de Bioquímica Facultad de Medicina Universidad Autónoma de Madrid microRNAs link the Germinal Center reaction and B cell lymphomagenesis Memoria presentada por la licenciada en Bioquímica Nahikari Bartolomé Izquierdo para optar al título de Doctor por la Universidad Autónoma de Madrid Directores de tesis: Dra. Almudena R. Ramiro Dra. Virginia García de Yébenes Este trabajo ha sido realizado en el laboratorio de Linfocitos B, en Centro Nacional de Investigaciones Cardiovasculares (CNIC) Madrid, 2016 ABSTRACT Germinal centers (GC) are microstructures where B cells that have been activated by antigen undergo a developmental program that involves the somatic diversification of their immunoglobulin genes and intense proliferative activity coupled to apoptotic selective events. As a result, from GCs emerge plasma cells and memory B cells that bear immunoglobulins with higher affinity for the initi- ating antigen and with more versatile effector functions. While ensuring a competent humoral im- mune response, GCs are also a site particularly prone to neoplastic transformation. Indeed, the vast majority of human lymphomas arise from GC-experienced B cells. MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate gene expression post- transcriptionally. In the past years miRNAs have arisen as key regulators of the immune function, however little is known of their role in GC B cells. In this work, we have identified miR-217 and miR- 28 as GC microRNAs and have explored their functional impact in GC events and in B cell lym- phomagenesis.