Selecting the Most Appropriate Time Points to Profile in High-Throughput Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Core Transcriptional Regulatory Circuitries in Cancer
Oncogene (2020) 39:6633–6646 https://doi.org/10.1038/s41388-020-01459-w REVIEW ARTICLE Core transcriptional regulatory circuitries in cancer 1 1,2,3 1 2 1,4,5 Ye Chen ● Liang Xu ● Ruby Yu-Tong Lin ● Markus Müschen ● H. Phillip Koeffler Received: 14 June 2020 / Revised: 30 August 2020 / Accepted: 4 September 2020 / Published online: 17 September 2020 © The Author(s) 2020. This article is published with open access Abstract Transcription factors (TFs) coordinate the on-and-off states of gene expression typically in a combinatorial fashion. Studies from embryonic stem cells and other cell types have revealed that a clique of self-regulated core TFs control cell identity and cell state. These core TFs form interconnected feed-forward transcriptional loops to establish and reinforce the cell-type- specific gene-expression program; the ensemble of core TFs and their regulatory loops constitutes core transcriptional regulatory circuitry (CRC). Here, we summarize recent progress in computational reconstitution and biologic exploration of CRCs across various human malignancies, and consolidate the strategy and methodology for CRC discovery. We also discuss the genetic basis and therapeutic vulnerability of CRC, and highlight new frontiers and future efforts for the study of CRC in cancer. Knowledge of CRC in cancer is fundamental to understanding cancer-specific transcriptional addiction, and should provide important insight to both pathobiology and therapeutics. 1234567890();,: 1234567890();,: Introduction genes. Till now, one critical goal in biology remains to understand the composition and hierarchy of transcriptional Transcriptional regulation is one of the fundamental mole- regulatory network in each specified cell type/lineage. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer
ORIGINAL RESEARCH published: 15 April 2021 doi: 10.3389/fonc.2021.566539 Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer Qinglian He 1, Ziqi Li 1, Jinbao Yin 1, Yuling Li 2, Yuting Yin 1, Xue Lei 1 and Wei Zhu 1* 1 Department of Pathology, Guangdong Medical University, Dongguan, China, 2 Department of Pathology, Dongguan People’s Hospital, Southern Medical University, Dongguan, China Background: Colorectal cancer (CRC) is a common malignant solid tumor with an extremely low survival rate after relapse. Previous investigations have shown that autophagy possesses a crucial function in tumors. However, there is no consensus on the value of autophagy-associated genes in predicting the prognosis of CRC patients. Edited by: This work screens autophagy-related markers and signaling pathways that may Fenglin Liu, Fudan University, China participate in the development of CRC, and establishes a prognostic model of CRC Reviewed by: based on autophagy-associated genes. Brian M. Olson, Emory University, United States Methods: Gene transcripts from the TCGA database and autophagy-associated gene Zhengzhi Zou, data from the GeneCards database were used to obtain expression levels of autophagy- South China Normal University, China associated genes, followed by Wilcox tests to screen for autophagy-related differentially Faqing Tian, Longgang District People's expressed genes. Then, 11 key autophagy-associated genes were identified through Hospital of Shenzhen, China univariate and multivariate Cox proportional hazard regression analysis and used to Yibing Chen, Zhengzhou University, China establish prognostic models. Additionally, immunohistochemical and CRC cell line data Jian Tu, were used to evaluate the results of our three autophagy-associated genes EPHB2, University of South China, China NOL3, and SNAI1 in TCGA. -

Familial Cortical Myoclonus Caused by Mutation in NOL3 by Jonathan Foster Rnsseil DISSERTATION Submitted in Partial Satisfaction
Familial Cortical Myoclonus Caused by Mutation in NOL3 by Jonathan Foster Rnsseil DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences in the Copyright 2013 by Jonathan Foster Russell ii I dedicate this dissertation to Mom and Dad, for their adamantine love and support iii No man has earned the right to intellectual ambition until he has learned to lay his course by a star which he has never seen—to dig by the divining rod for springs which he may never reach. In saying this, I point to that which will make your study heroic. For I say to you in all sadness of conviction, that to think great thoughts you must be heroes as well as idealists. Only when you have worked alone – when you have felt around you a black gulf of solitude more isolating than that which surrounds the dying man, and in hope and in despair have trusted to your own unshaken will – then only will you have achieved. Thus only can you gain the secret isolated joy of the thinker, who knows that, a hundred years after he is dead and forgotten, men who never heard of him will be moving to the measure of his thought—the subtile rapture of a postponed power, which the world knows not because it has no external trappings, but which to his prophetic vision is more real than that which commands an army. -Oliver Wendell Holmes, Jr. iv ACKNOWLEDGMENTS I am humbled by the efforts of many, many others who were essential for this work. -

Supplemental Methods Proband and Control Lung Samples Lung Sections from Both the Proband and Age-Matched Controls Were Obtained
Supplementary material J Med Genet Supplemental Methods Proband and Control Lung Samples Lung sections from both the proband and age-matched controls were obtained in the form of fixed formalin paraffin embedded (FFPE) samples. Control lung samples were obtained from the BRINDL biorepository at the University of Rochester(1). RNA expression analyses RNA was obtained for mRNA expression studies using the RNeasy FFPE kit (Qiagen) according to manufacturer’s instructions. cDNA was then generated using iScrpit kit (biorad), and quantitative PCR was done as previously published (2). Primers for qPCR are listed in supplemental table 1. Chromatin Immunoprecipitation of FFPE for Fixed Formalin Paraffin Embedded samples Chromatin immunoprecipitation was done on FFPE sections using a protocol modified from Fanelli and colleagues, Nature Methods, 2011(3) Samples were deparaffinized by incubating 10uM sections in 1ml of Xylene for 10 minutes at room temperature. The tissue was then pelleted at 17,000 x gravity for 3 min at room temperature. This process was repeated 3 times. Samples were then rehydrated by incubating with 1 ml of 100% Ethanol for 10 minutes at RT. Cells were pelleted and resuspended in progressively increasing percentage of water as follows: 95% ethanol, 70% ethanol, 50% ethanol, 20% ethanol. The sample was then resuspended in 1x PBS and the tissue dissociated by sonicating with a Bioruptor (Diagnode, NJ USA) for 30 seconds on the medium setting. Cells then resuspended in Collagenase digestion buffer and treated with collagenase A (Roche 11 088 785 103) to a final concentration of 2mg/ml and digested at 37 C for 45 minutes. -

Daneshshahab.Pdf (3.352Mb)
BMP SIGNALING THROUGH BMPR1A IS REQUIRED FOR ESTABLISHMENT OF PANCREATIC LATERALITY APPROVED BY SUPERVISORY COMMITTEE Ondine Cleaver, Ph.D. Melanie H. Cobb, Ph.D. Raymond J. MacDonald, Ph.D. Eric N. Olson, Ph.D. THIS DISSERTATION IS DEDICATED TO MY FAMILY, PAST, PRESENT AND FUTURE BMP SIGNALING THROUGH BMPR1A IS REQUIRED FOR ESTABLISHMENT OF PANCREATIC LATERALITY by SHAHAB SHAUN MALEKPOUR DANESH DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas May, 2009 Copyright by SHAHAB SHAUN MALEKPOUR DANESH, 2009 All Rights Reserved ACKNOWLEDGEMENTS I would like to thank my thesis advisor, Dr. Ondine Cleaver for her guidance and support. Ondine accepted me into her lab as a fifth year graduate student who had already been in three labs and had no background in developmental biology or mouse work. Her undying guidance, motivation and enthusiasm for science have allowed me to grow as a scientist and complete my thesis in two short years. I would like to thank my committee members Dr. Melanie Cobb, Dr. Eric Olson, and especially my thesis chair, Dr. Raymond MacDonald. They have provided invaluable suggestions and guidance that have facilitated my project that has allowed me to complete a substantial amount of work in a short time. I would like to thank the members of the Cleaver lab for providing a collaborative environment for science. Aly Villasenor was a great resource for exchange of valuable scientific ideas. -

Gene Regulation Is Governed by a Core Network in Hepatocellular Carcinoma Zuguang Gu, Chenyu Zhang* and Jin Wang*
Gu et al. BMC Systems Biology 2012, 6:32 http://www.biomedcentral.com/1752-0509/6/32 RESEARCH ARTICLE Open Access Gene regulation is governed by a core network in hepatocellular carcinoma Zuguang Gu, Chenyu Zhang* and Jin Wang* Abstract Background: Hepatocellular carcinoma (HCC) is one of the most lethal cancers worldwide, and the mechanisms that lead to the disease are still relatively unclear. However, with the development of high-throughput technologies it is possible to gain a systematic view of biological systems to enhance the understanding of the roles of genes associated with HCC. Thus, analysis of the mechanism of molecule interactions in the context of gene regulatory networks can reveal specific sub-networks that lead to the development of HCC. Results: In this study, we aimed to identify the most important gene regulations that are dysfunctional in HCC generation. Our method for constructing gene regulatory network is based on predicted target interactions, experimentally-supported interactions, and co-expression model. Regulators in the network included both transcription factors and microRNAs to provide a complete view of gene regulation. Analysis of gene regulatory network revealed that gene regulation in HCC is highly modular, in which different sets of regulators take charge of specific biological processes. We found that microRNAs mainly control biological functions related to mitochondria and oxidative reduction, while transcription factors control immune responses, extracellular activity and the cell cycle. On the higher level of gene regulation, there exists a core network that organizes regulations between different modules and maintains the robustness of the whole network. There is direct experimental evidence for most of the regulators in the core gene regulatory network relating to HCC. -

Prenatal Testing Requisition Form
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 PRENATAL COMPREHENSIVE REQUISITION FORM PATIENT INFORMATION NAME (LAST,FIRST, MI): DATE OF BIRTH (MM/DD/YY): HOSPITAL#: ACCESSION#: REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: SAMPLE INFORMATION CLINICAL INDICATION FETAL SPECIMEN TYPE Pregnancy at risk for specific genetic disorder DATE OF COLLECTION: (Complete FAMILIAL MUTATION information below) Amniotic Fluid: cc AMA PERFORMING PHYSICIAN: CVS: mg TA TC Abnormal Maternal Screen: Fetal Blood: cc GESTATIONAL AGE (GA) Calculation for AF-AFP* NTD TRI 21 TRI 18 Other: SELECT ONLY ONE: Abnormal NIPT (attach report): POC/Fetal Tissue, Type: TRI 21 TRI 13 TRI 18 Other: Cultured Amniocytes U/S DATE (MM/DD/YY): Abnormal U/S (SPECIFY): Cultured CVS GA ON U/S DATE: WKS DAYS PARENTAL BLOODS - REQUIRED FOR CMA -OR- Maternal Blood Date of Collection: Multiple Pregnancy Losses LMP DATE (MM/DD/YY): Parental Concern Paternal Blood Date of Collection: Other Indication (DETAIL AND ATTACH REPORT): *Important: U/S dating will be used if no selection is made. Name: Note: Results will differ depending on method checked. Last Name First Name U/S dating increases overall screening performance. Date of Birth: KNOWN FAMILIAL MUTATION/DISORDER SPECIFIC PRENATAL TESTING Notice: Prior to ordering testing for any of the disorders listed, you must call the lab and discuss the clinical history and sample requirements with a genetic counselor. -
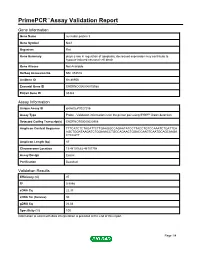
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name nucleolar protein 3 Gene Symbol Nol3 Organism Rat Gene Summary plays a role in regulation of apoptosis; decreased expression may contribute to hypoxia-induced neuronal cell death Gene Aliases Not Available RefSeq Accession No. NM_053516 UniGene ID Rn.86956 Ensembl Gene ID ENSRNOG00000015588 Entrez Gene ID 85383 Assay Information Unique Assay ID qRnoCEP0027256 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000020908 Amplicon Context Sequence TTTCATCTCTAGATTCTTGAAGGCCAGAATATCCTTACCTGTCCAAATCTGATTCA AGCTGGATAAGATCTGGAAACCTGCCAGAACTGGACCAAGTCAATGCAGCAAGA CTCCATT Amplicon Length (bp) 87 Chromosome Location 19:48101682-48101798 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9998 cDNA Cq 22.33 cDNA Tm (Celsius) 80 gDNA Cq 26.04 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Nol3, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Clinical and Molecular Investigation of Rare
CLINICAL AND MOLECULAR INVESTIGATION OF RARE CONGENITAL DEFECTS OF THE PALATE RIMANTE SESELGYTE A thesis submitted for the degree of Doctor of Philosophy to University College London August 2019 page Title 1 DECLARATION I, Rimante Seselgyte, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Signed……………………….. Declaration 3 ABSTRACT Cleft palate (CP) affects around 1/1500 live births and, along with cleft lip, is one of the most common forms of birth defect. The studies presented here focus on unusual defects of the palate, especially to understand better the rarely reported but surprisingly common condition called submucous cleft palate (SMCP). The frequency and consequences of SMCP from a surgical perspective were first investigated based on the caseload of the North Thames Cleft Service at Great Ormond Street Hospital and St Andrew's Centre, Broomfield Hospital, Mid Essex Hospitals Trust. It was previously reported that up to 80% of individuals with unrepaired SMCP experience speech difficulties as a consequence of velopharyngeal insufficiency (VPI). Attempted repair of the palatal defect can sometimes give poor results, so controversies still exist about the correct choice of surgical technique to use. Over 23 years, 222 patients at The North Thames Cleft Service underwent operations to manage SMCP. Nearly half of them (42.8%) were diagnosed with 22q11.2 deletion syndrome (22q11.2 DS). The first operation was palate repair, with an exception of one case, followed by a second surgical intervention required in approximately half of the patients. -

Medical Relevance of Protein-Truncating Variants Across 337,205 Individuals in the UK Biobank Study
ARTICLE DOI: 10.1038/s41467-018-03910-9 OPEN Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study Christopher DeBoever1,2, Yosuke Tanigawa 1, Malene E. Lindholm3, Greg McInnes1, Adam Lavertu1, Erik Ingelsson4, Chris Chang3, Euan A. Ashley 5, Carlos D. Bustamante1,2, Mark J. Daly 6,7 & Manuel A. Rivas1 Protein-truncating variants can have profound effects on gene function and are critical for 1234567890():,; clinical genome interpretation and generating therapeutic hypotheses, but their relevance to medical phenotypes has not been systematically assessed. Here, we characterize the effect of 18,228 protein-truncating variants across 135 phenotypes from the UK Biobank and find 27 associations between medical phenotypes and protein-truncating variants in genes outside the major histocompatibility complex. We perform phenome-wide analyses and directly measure the effect in homozygous carriers, commonly referred to as “human knockouts,” across medical phenotypes for genes implicated as being protective against disease or associated with at least one phenotype in our study. We find several genes with strong pleiotropic or non-additive effects. Our results illustrate the importance of protein-truncating variants in a variety of diseases. 1 Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University, Stanford, CA 94305, USA. 3 Grail, Inc., 1525 O’Brien Drive, Menlo Park, CA 94025, USA. 4 Division of Cardiovascular Medicine, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA. 5 Department of Medicine, School of Medicine, Stanford University, Stanford, CA 94305, USA. 6 Analytical and Translational Genetics Unit, Boston, MA 02114, USA. -

Wppi: Weighting Protein-Protein Interactions
Package ‘wppi’ September 23, 2021 Type Package Title Weighting protein-protein interactions Version 1.1.2 Description Protein-protein interaction data is essential for omics data analysis and modeling. Database knowledge is general, not specific for cell type, physiological condition or any other context determining which connections are functional and contribute to the signaling. Functional annotations such as Gene Ontology and Human Phenotype Ontology might help to evaluate the relevance of interactions. This package predicts functional relevance of protein-protein interactions based on functional annotations such as Human Protein Ontology and Gene Ontology, and prioritizes genes based on network topology, functional scores and a path search algorithm. License MIT + file LICENSE URL https://github.com/AnaGalhoz37/wppi BugReports https://github.com/AnaGalhoz37/wppi/issues biocViews GraphAndNetwork, Network, Pathways, Software, GeneSignaling, GeneTarget, SystemsBiology, Transcriptomics, Annotation Encoding UTF-8 VignetteBuilder knitr Depends R(>= 4.1) Imports dplyr, igraph, logger, methods, magrittr, Matrix, OmnipathR(>= 2.99.8), progress, purrr, rlang, RCurl, stats, tibble, tidyr Suggests knitr, testthat, rmarkdown RoxygenNote 7.1.1 NeedsCompilation no git_url https://git.bioconductor.org/packages/wppi git_branch master git_last_commit 6aa91be git_last_commit_date 2021-07-15 1 2 common_neighbors Date/Publication 2021-09-23 Author Ana Galhoz [cre, aut] (<https://orcid.org/0000-0001-7402-5292>), Denes Turei [aut] (<https://orcid.org/0000-0002-7249-9379>), Michael P. Menden [aut] (<https://orcid.org/0000-0003-0267-5792>), Albert Krewinkel [ctb, cph] (pagebreak Lua filter) Maintainer Ana Galhoz <[email protected]> R topics documented: common_neighbors . .2 count_genes . .3 filter_annot_with_network . .4 functional_annot . .5 graph_from_op . .6 in_omnipath . .6 prioritization_genes . .7 process_annot .