TNF-Stimulated MAP Kinase Activation Mediated by a Rho Family Gtpase Signaling Pathway
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TAK1 Mediates Convergence of Cellular Signals for Death and Survival
Apoptosis (2019) 24:3–20 https://doi.org/10.1007/s10495-018-1490-7 REVIEW TAK1 mediates convergence of cellular signals for death and survival Sabreena Aashaq1 · Asiya Batool1 · Khurshid I. Andrabi1 Published online: 4 October 2018 © Springer Science+Business Media, LLC, part of Springer Nature 2018 Abstract TGF-β activated kinase 1, a MAPK kinase kinase family serine threonine kinase has been implicated in regulating diverse range of cellular processes that include embryonic development, differentiation, autophagy, apoptosis and cell survival. TAK1 along with its binding partners TAB1, TAB2 and TAB3 displays a complex pattern of regulation that includes serious crosstalk with major signaling pathways including the C-Jun N-terminal kinase (JNK), p38 MAPK, and I-kappa B kinase complex (IKK) involved in establishing cellular commitments for death and survival. This review also highlights how TAK1 orchestrates regulation of energy homeostasis via AMPK and its emerging role in influencing mTORC1 pathway to regulate death or survival in tandem. Keywords Apoptosis · Autophagy · Cytokine · Inflammatory · Smad TAK1, a multifunctional kinase regulate a wide array of downstream cellular responses [2, 7, 8]. Various branches of MAP kinase pathways including Transforming growth factor-β is a versatile cytokine, regu- the extracellular signal regulated kinase (Erk) ½ [3, 9], p38 lating a wide variety of intracellular signaling pathways. MAPK [10, 11], c-Jun N-Terminal kinase (JNK) [12, 13], The Smad dependent signaling pathway is conventionally phosphatidylinositol-3-kinase/AKT pathway [14, 15] and acknowledged as the traditional pathway promoted by TGF- Rho-like GTPase [16, 17] signaling pathways are included β1 [1]. However, the Smad dependent signaling pathway among the Smad independent pathways. -

High Expression of LINC01268 Is Positively Associated with Hepatocellular Carcinoma Progression Via Regulating MAP3K7
OncoTargets and Therapy Dovepress open access to scientific and medical research Open Access Full Text Article ORIGINAL RESEARCH High Expression of LINC01268 is Positively Associated with Hepatocellular Carcinoma Progression via Regulating MAP3K7 This article was published in the following Dove Press journal: OncoTargets and Therapy Xiuli Jin,1 Weixin Fu,2 Dan Li,1 Objective: As one of the most common neoplastic diseases, hepatocellular carcinoma Ningning Wang,1 Jiayu Chen,1 (HCC) has a high morbidity and mortality, which seriously threatens human health and Zilu Zeng,1 Jiaqi Guo,1 Hao places a heavy burden on society and medical care. At present, effective early diagnosis, Liu,3 Xinping Zhong,3 Hu prognosis and treatment of HCC are limited. Altered gene expression patterns of lncRNA are Peng,4 Xin Yu,5 Jing Sun,1 associated with the occurrence, development and prognosis of various malignancies, includ- ing HCC. The aim of this study was to investigate the correlation between the expression of Xinhe Zhang,1 Xue Wang,1 LINC01268 and HCC, and to elucidate the potential underlying molecular mechanism. Beibei Xu,1 Yingbo Lin,6 4 Methods: Expression level and localization of LINC01268 in human liver cancer cells and Jianping Liu, Claudia HCC tissues were investigated using RT-qPCR and fluorescent in situ hybridization (FISH), 7 1 Kutter, Yiling Li respectively. Correlation of expression levels of LINC01268 and MAP3K7 with differentia- 1Department of Gastroenterology, First tion and poor overall patient survival of HCC were analyzed using in house collected and AffiliatedHospital of China Medical publicly available HCC tissue data. RT-qPCR and Western blot were applied to inspect the University, Shenyang, 110001, People’s Republic of China; 2Science Experiment effects of depletion and overexpression of LINC01268 on MAP3K7 expression. -

MAP Kinase Kinase Kinase, MEKK, MAPKKK
MAP3K MAP kinase kinase kinase, MEKK, MAPKKK MAP3Ks (Mitogen-activated protein kinase kinase kinases), the top components of MAPK cascades, provide specificity for stimulus-dependent activation of MAP2K-MAPK pathways through unique protein-protein interactions and phosphorylation of signaling effectors. The MAP3Ks are highly divergent in gene numbers and structure, including TAK1, ASK1, A-Raf and C-Raf. MAPK system is a three-step sequential phosphorylation cascade which is composed of MAPK, MAP2K, and MAP3K. ERK, p38 MAPK, and JNK, which are known to be activated by mechanical stimuli, belong to the MAPK family. MAP3Ks function as “platforms to integrate MAPK signaling, and activation of multiple MAP3Ks provides the spatiotemporal regulation of the MAPK pathways, which induces a wide range of physiological responses required for determining cell fate, such as cytokine production, survival, differentiation and apoptosis”. www.MedChemExpress.com 1 MAP3K Inhibitors 5Z-7-Oxozeaenol ASK1-IN-2 (FR148083; L783279; LL-Z 1640-2) Cat. No.: HY-12686 Cat. No.: HY-131969 5Z-7-Oxozeaenol is a natural anti-protozoan ASK1-IN-2 is a potent and orally active inhibitor compound from fungal origin, acting as a potent of apoptosis signal-regulating kinase 1 (ASK1), irreversible and selective inhibitor of TAK1 and with an IC50 of 32.8 nM. ASK1-IN-2 can be used for VEGF-R2, with IC50s of 8 nM and 52 nM, the research of ulcerative colitis. respectively. Purity: ≥99.0% Purity: 98.49% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 1 mg Size: 10 mM × 1 mL, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg Cot inhibitor-1 Cot inhibitor-2 Cat. -

MAP3K7 Antibody Purified Mouse Monoclonal Antibody Catalog # Ao2028a
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 MAP3K7 Antibody Purified Mouse Monoclonal Antibody Catalog # AO2028a Specification MAP3K7 Antibody - Product Information Application WB, E Primary Accession O43318 Reactivity Human Host Mouse Clonality Monoclonal Isotype IgG2a Calculated MW 67.2kDa KDa Description The protein encoded by this gene is a member of the serine/threonine protein kinase family. This kinase mediates the signaling transduction induced by TGF beta and morphogenetic protein (BMP), and controls a variety of cell functions including transcription regulation and apoptosis. In response to IL-1, this protein forms a kinase complex including TRAF6, MAP3K7P1/TAB1 and MAP3K7P2/TAB2; this complex is required for the activation of nuclear factor kappa B. This kinase can also activate MAPK8/JNK, MAP2K4/MKK4, and thus plays a role in the cell response to environmental stresses. Four alternatively spliced transcript variants encoding distinct isoforms have been reported. Immunogen Purified recombinant fragment of human MAP3K7 (AA: 471-579) expressed in E. Coli. Formulation Purified antibody in PBS with 0.05% sodium azide and 0.5% protein stabilizer MAP3K7 Antibody - Additional Information Gene ID 6885 Other Names Mitogen-activated protein kinase kinase kinase 7, 2.7.11.25, Transforming growth factor-beta-activated kinase 1, TGF-beta-activated kinase 1, MAP3K7, TAK1 Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Dilution WB~~1/500 - 1/2000 E~~1/10000 Storage Maintain refrigerated at 2-8°C for up to 6 months. -
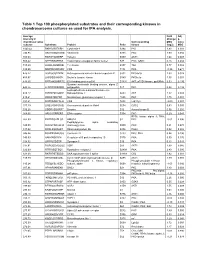
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S
crossmark Research Author’s Choice © 2016 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy*□S Rolf Turk‡§¶ʈ**, Jordy J. Hsiao¶, Melinda M. Smits¶, Brandon H. Ng¶, Tyler C. Pospisil‡§¶ʈ**, Kayla S. Jones‡§¶ʈ**, Kevin P. Campbell‡§¶ʈ**, and Michael E. Wright¶‡‡ Mutations in genes encoding components of the sar- The muscular dystrophies are hereditary diseases charac- colemmal dystrophin-glycoprotein complex (DGC) are re- terized primarily by the progressive degeneration and weak- sponsible for a large number of muscular dystrophies. As ness of skeletal muscle. Most are caused by deficiencies in such, molecular dissection of the DGC is expected to both proteins associated with the cell membrane (i.e. the sarco- reveal pathological mechanisms, and provides a biologi- lemma in skeletal muscle), and typical features include insta- cal framework for validating new DGC components. Es- bility of the sarcolemma and consequent death of the myofi- tablishment of the molecular composition of plasma- ber (1). membrane protein complexes has been hampered by a One class of muscular dystrophies is caused by mutations lack of suitable biochemical approaches. Here we present in genes that encode components of the sarcolemmal dys- an analytical workflow based upon the principles of pro- tein correlation profiling that has enabled us to model the trophin-glycoprotein complex (DGC). In differentiated skeletal molecular composition of the DGC in mouse skeletal mus- muscle, this structure links the extracellular matrix to the cle. We also report our analysis of protein complexes in intracellular cytoskeleton. -

REVIEW Gene Expression Profile of Human Thyroid Cancer in Relation To
R91 REVIEW Gene expression profile of human thyroid cancer in relation to its mutational status Dagmara Rusinek, Sylwia Szpak-Ulczok and Barbara Jarzab Department of Nuclear Medicine and Endocrine Oncology, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Gliwice Branch, Wybrzeze Armii Krajowej 15, 44-101 Gliwice, Poland (Correspondence should be addressed to B Jarzab; Email: [email protected]) Abstract This review describes the gene expression profile changes associated with the presence of different mutations that contribute to thyroid cell carcinogenesis. The results are discussed in the context of thyroid cancer biology and of the implications for disease prognosis, while the diagnostic aspect has been omitted. For papillary thyroid cancer (PTC), the most characteristic gene expression profile is associated with the presence of BRAF mutation. BRAF-associated PTC differ profoundly from RET/PTC or RAS-associated cancers. Simultaneously, they retain many characteristic gene expression features common for all PTCs, induced by the alternative mutations activating MAPK pathway. Although the difference between papillary and follicular thyroid cancer (FTC) is significant at the gene expression profile level, surprisingly, the RAS-related signature of FTC is not well specified. PAX8/peroxisome proliferator-activated receptor g (PPARg) rearrangements, which occur in FTC as an alternative to the RAS mutation, are associated with specific changes in gene expression. Furthermore, the difference between well-differentiated thyroid cancers and poorly differentiated and anaplastic thyroid cancers is mainly a reflection of tumor degree of differentiation and may not be attributed to the presence of characteristic mutations. Journal of Molecular Endocrinology (2011) 47, R91–R103 Introduction Transcriptome of papillary thyroid cancer Gene expression profiling of thyroid tumors has Papillary thyroid cancer develops directly from the expanded our knowledge of the molecular biology of thyroid cells and does not have a benign intermediate. -

Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action
International Journal of Molecular Sciences Review Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action Paweł Łukasik 1, Irena Baranowska-Bosiacka 2, Katarzyna Kulczycka 3 and Izabela Gutowska 1,* 1 Department of Medical Chemistry, Pomeranian Medical University in Szczecin, Powstancow Wlkp. 72 Av., 70-111 Szczecin, Poland; [email protected] 2 Department of Biochemistry, Pomeranian Medical University in Szczecin, Powstancow Wlkp. 72 Av., 70-111 Szczecin, Poland; [email protected] 3 Department of Pharmaceutical Chemistry, Pomeranian Medical University in Szczecin, Powstancow Wlkp. 72 Av., 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Abstract: Recent studies on cyclin-dependent kinase (CDK) inhibitors have revealed that small molecule drugs have become very attractive for the treatment of cancer and neurodegenerative disorders. Most CDK inhibitors have been developed to target the ATP binding pocket. However, CDK kinases possess a very similar catalytic domain and three-dimensional structure. These features make it difficult to achieve required selectivity. Therefore, inhibitors which bind outside the ATP binding site present a great interest in the biomedical field, both from the fundamental point of view and for the wide range of their potential applications. This review tries to explain whether the ATP competitive inhibitors are still an option for future research, and highlights alternative approaches to discover more selective and potent small molecule inhibitors. Keywords: cyclin-dependent kinase inhibitors; cancer; cell cycle; CDKs; CDK inhibitors Citation: Łukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types 1. Cyclin-Dependent Kinases (CDKs) and Their Mechanism of Action. -

Inhibition of ERK 1/2 Kinases Prevents Tendon Matrix Breakdown Ulrich Blache1,2,3, Stefania L
www.nature.com/scientificreports OPEN Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown Ulrich Blache1,2,3, Stefania L. Wunderli1,2,3, Amro A. Hussien1,2, Tino Stauber1,2, Gabriel Flückiger1,2, Maja Bollhalder1,2, Barbara Niederöst1,2, Sandro F. Fucentese1 & Jess G. Snedeker1,2* Tendon extracellular matrix (ECM) mechanical unloading results in tissue degradation and breakdown, with niche-dependent cellular stress directing proteolytic degradation of tendon. Here, we show that the extracellular-signal regulated kinase (ERK) pathway is central in tendon degradation of load-deprived tissue explants. We show that ERK 1/2 are highly phosphorylated in mechanically unloaded tendon fascicles in a vascular niche-dependent manner. Pharmacological inhibition of ERK 1/2 abolishes the induction of ECM catabolic gene expression (MMPs) and fully prevents loss of mechanical properties. Moreover, ERK 1/2 inhibition in unloaded tendon fascicles suppresses features of pathological tissue remodeling such as collagen type 3 matrix switch and the induction of the pro-fbrotic cytokine interleukin 11. This work demonstrates ERK signaling as a central checkpoint to trigger tendon matrix degradation and remodeling using load-deprived tissue explants. Tendon is a musculoskeletal tissue that transmits muscle force to bone. To accomplish its biomechanical function, tendon tissues adopt a specialized extracellular matrix (ECM) structure1. Te load-bearing tendon compart- ment consists of highly aligned collagen-rich fascicles that are interspersed with tendon stromal cells. Tendon is a mechanosensitive tissue whereby physiological mechanical loading is vital for maintaining tendon archi- tecture and homeostasis2. Mechanical unloading of the tissue, for instance following tendon rupture or more localized micro trauma, leads to proteolytic breakdown of the tissue with severe deterioration of both structural and mechanical properties3–5. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

SNP Gene Chr* Region P Value Odd Ratios Minor Allele Major Allele Rs11184708 PRMT6 1 Upstream 6.447× 10−13 6.149 T a Rs108025
Supplementary Table S1. Detailed information on scrub typhus-related candidate SNPs with a p value < 1 × 10−4. Odd Minor Major SNP Gene Chr* Region p value Ratios Allele Allele rs11184708 PRMT6 1 upstream 6.447× 10−13 6.149 T A rs10802595 RYR2 1 intron 0.00008738 2.593 A G downstream, intron, rs401974 LINC00276,LOC100506474 2 0.0000769 0.3921 T C upstream rs1445126 MIR4757,NT5C1B 2 upstream 0.00004819 3.18 A G LOC101930107,MIR4435-1,PLGLB rs62140478 2 downstream, upstream 7.404 × 10−8 9.708 T C 2 rs35890165 CPS1,ERBB4 2 downstream 0.00003952 0.373 A G rs34599430 ZNF385D,ZNF385D-AS2 3 intron, upstream 0.00002317 2.767 G A rs6809058 RBMS3,TGFBR2 3 downstream, upstream 0.00002507 3.094 G A rs3773683 SIDT1 3 intron 0.00006635 0.3543 C T rs11727383 CRMP1,EVC 4 intron 0.0000803 0.3459 A G rs17338338 NUDT12,RAB9BP1 5 upstream 0.00006987 0.3746 G A rs2059950 DTWD2,LOC102467225 5 downstream 0.000008739 2.911 G A rs72663337 DTWD2,LOC102467225 5 downstream 0.00009856 2.566 T C rs6882516 LSM11 5 UTR-3 0.00007553 2.968 A C rs76949230 TENM2 5 intron 0.00006305 3.048 G C rs3804468 LY86,LY86-AS1 6 intron 0.00003155 0.202 C T rs3778337 DSP 6 exon,intron 0.00007311 0.3897 G A rs16883596 MAP3K7,MIR4643 6 upstream 0.00005186 4.274 A G rs35144103 CCT6P3,ZNF92 7 downstream, upstream 0.00004885 3.422 A G rs13244090 LOC407835,TPI1P2 7 downstream, upstream 0.00004505 2.638 G A rs17167553 LRGUK 7 missense 0.00008788 2.75 T G rs6583826 IDE,KIF11 10 upstream 0.00006894 0.2846 G A rs10769111 LOC221122,PRDM11 11 downstream, upstream 0.0000619 0.3816 T G rs10848921 -

Targeting the Hsp90-Cdc37-Client Protein Interaction to Disrupt Hsp90 Chaperone Machinery Ting Li1, Hu-Lin Jiang2, Yun-Guang Tong3,4 and Jin-Jian Lu1*
Li et al. Journal of Hematology & Oncology (2018) 11:59 https://doi.org/10.1186/s13045-018-0602-8 REVIEW Open Access Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery Ting Li1, Hu-Lin Jiang2, Yun-Guang Tong3,4 and Jin-Jian Lu1* Abstract Heat shock protein 90 (Hsp90) is a critical molecular chaperone protein that regulates the folding, maturation, and stability of a wide variety of proteins. In recent years, the development of Hsp90-directed inhibitors has grown rapidly, and many of these inhibitors have entered clinical trials. In parallel, the functional dissection of the Hsp90 chaperone machinery has highlighted the activity disruption of Hsp90 co-chaperone as a potential target. With the roles of Hsp90 co-chaperones being elucidated, cell division cycle 37 (Cdc37), a ubiquitous co-chaperone of Hsp90 that directs the selective client proteins into the Hsp90 chaperone cycle, shows great promise. Moreover, the Hsp90-Cdc37-client interaction contributes to the regulation of cellular response and cellular growth and is more essential to tumor tissues than normal tissues. Herein, we discuss the current understanding of the clients of Hsp90-Cdc37, the interaction of Hsp90-Cdc37-client protein, and the therapeutic possibilities of targeting Hsp90-Cdc37-client protein interaction as a strategy to inhibit Hsp90 chaperone machinery to present new insights on alternative ways of inhibiting Hsp90 chaperone machinery. Keywords: Hsp90 chaperone machinery, Cdc37, Kinase client, Protein interaction Background chloroplast HSP90C, mitochondrial TNFR-associated protein, Heat shock protein 90 (Hsp90) is a critically conserved and bacterial high-temperature protein G [2, 8]. In this protein and one of the major molecular chaperones within review,weusethetermHsp90torefertotheseHsp90 eukaryotic cells [1].