A Fatal Case of Valproate-Induced Hyperammonemic Encephalopathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Migraine Prophylaxis: Which Drugs Work and Which Ones Don't
Migraine Prophylaxis: Which Drugs Work and Which Ones Don’t Hans-Christoph Diener, MD Department of Neurology, University Hospital Essen, Essen, Germany. J Gen Intern Med 28(9):1125–6 investigated in two properly powered and conducted studies DOI: 10.1007/s11606-013-2469-2 and found not to be effective compared to placebo.3, 4 Adding © Society of General Internal Medicine 2013 data from poorly controlled and underpowered studies in a meta-analysis, as in this paper, gives the wrong impression that he paper by Shamliyan et al.1 in this issue of JGIM is a very nimodipine is as effective in migraine prevention as propran- T important contribution to headache research. The authors olol. Another example illustrated in this meta-analysis involves conducted a systematic literature review of drug treatment for the gabapentin. Most of the published trials are poorly conducted, underpowered, or have manipulated statistical analyses, such as prevention of episodic migraine. They analysed randomised 5 controlled trials (RCTs) and performed meta-analyses where modified intention-to-treat analyses. The meta-analysis in this appropriate. The two important outcomes examined in the paper indicated possible efficacy. Yet, in the time between the analysis are a ≥ 50 % reduction in migraine frequency and submission and publication of this paper, a recent well-powered adverse events leading to treatment discontinuation. dose finding trial was published investigating four doses of gabapentin as compared with placebo for migraine prevention. 6 What are the strengths of this paper? This trial showed no benefit for gabapentin. Another problem inherent in meta-analyses that this paper The authors are experts in this kind of analysis. -

Optum Essential Health Benefits Enhanced Formulary PDL January
PENICILLINS ketorolac tromethamineQL GENERIC mefenamic acid amoxicillin/clavulanate potassium nabumetone amoxicillin/clavulanate potassium ER naproxen January 2016 ampicillin naproxen sodium ampicillin sodium naproxen sodium CR ESSENTIAL HEALTH BENEFITS ampicillin-sulbactam naproxen sodium ER ENHANCED PREFERRED DRUG LIST nafcillin sodium naproxen DR The Optum Preferred Drug List is a guide identifying oxacillin sodium oxaprozin preferred brand-name medicines within select penicillin G potassium piroxicam therapeutic categories. The Preferred Drug List may piperacillin sodium/ tazobactam sulindac not include all drugs covered by your prescription sodium tolmetin sodium drug benefit. Generic medicines are available within many of the therapeutic categories listed, in addition piperacillin sodium/tazobactam Fenoprofen Calcium sodium to categories not listed, and should be considered Meclofenamate Sodium piperacillin/tazobactam as the first line of prescribing. Tolmetin Sodium Amoxicillin/Clavulanate Potassium LOW COST GENERIC PREFERRED For benefit coverage or restrictions please check indomethacin your benefit plan document(s). This listing is revised Augmentin meloxicam periodically as new drugs and new prescribing LOW COST GENERIC naproxen kit information becomes available. It is recommended amoxicillin that you bring this list of medications when you or a dicloxacillin sodium CARDIOVASCULAR covered family member sees a physician or other penicillin v potassium ACE-INHIBITORS healthcare provider. GENERIC QUINOLONES captopril ANTI-INFECTIVES -

Neurontin (Gabapentin)
Texas Prior Authorization Program Clinical Criteria Drug/Drug Class Gabapentin Clinical Criteria Information Included in this Document Neurontin (gabapentin) • Drugs requiring prior authorization: the list of drugs requiring prior authorization for this clinical criteria • Prior authorization criteria logic: a description of how the prior authorization request will be evaluated against the clinical criteria rules • Logic diagram: a visual depiction of the clinical criteria logic • Supporting tables: a collection of information associated with the steps within the criteria (diagnosis codes, procedure codes, and therapy codes); provided when applicable • References: clinical publications and sources relevant to this clinical criteria Note: Click the hyperlink to navigate directly to that section. Gralise (gabapentin Extended Release) • Drugs requiring prior authorization: the list of drugs requiring prior authorization for this clinical criteria • Prior authorization criteria logic: a description of how the prior authorization request will be evaluated against the clinical criteria rules • Logic diagram: a visual depiction of the clinical criteria logic • Supporting tables: a collection of information associated with the steps within the criteria (diagnosis codes, procedure codes, and therapy codes); provided when applicable • References: clinical publications and sources relevant to this clinical criteria Note: Click the hyperlink to navigate directly to that section. March 29, 2019 Copyright © 2019 Health Information Designs, LLC 1 Horizant -

Migraine Headache Prophylaxis Hien Ha, Pharmd, and Annika Gonzalez, MD, Christus Santa Rosa Family Medicine Residency Program, San Antonio, Texas
Migraine Headache Prophylaxis Hien Ha, PharmD, and Annika Gonzalez, MD, Christus Santa Rosa Family Medicine Residency Program, San Antonio, Texas Migraines impose significant health and financial burdens. Approximately 38% of patients with episodic migraines would benefit from preventive therapy, but less than 13% take prophylactic medications. Preventive medication therapy reduces migraine frequency, severity, and headache-related distress. Preventive therapy may also improve quality of life and prevent the progression to chronic migraines. Some indications for preventive therapy include four or more headaches a month, eight or more headache days a month, debilitating headaches, and medication- overuse headaches. Identifying and managing environmental, dietary, and behavioral triggers are useful strategies for preventing migraines. First-line med- ications established as effective based on clinical evidence include divalproex, topiramate, metoprolol, propranolol, and timolol. Medications such as ami- triptyline, venlafaxine, atenolol, and nadolol are probably effective but should be second-line therapy. There is limited evidence for nebivolol, bisoprolol, pindolol, carbamazepine, gabapentin, fluoxetine, nicardipine, verapamil, nimodipine, nifedipine, lisinopril, and candesartan. Acebutolol, oxcarbazepine, lamotrigine, and telmisartan are ineffective. Newer agents target calcitonin gene-related peptide pain transmission in the migraine pain pathway and have recently received approval from the U.S. Food and Drug Administration; how- ever, more studies of long-term effectiveness and adverse effects are needed. The complementary treatments petasites, feverfew, magnesium, and riboflavin are probably effective. Nonpharmacologic therapies such as relaxation training, thermal biofeedback combined with relaxation training, electromyographic feedback, and cognitive behavior therapy also have good evidence to support their use in migraine prevention. (Am Fam Physician. 2019; 99(1):17-24. -

Original Article Comparison of Therapeutic Effects of Two Ccbs on Glaucoma and Analysis of Their Possible Mechanisms
Int J Clin Exp Med 2017;10(7):10560-10564 www.ijcem.com /ISSN:1940-5901/IJCEM0056272 Original Article Comparison of therapeutic effects of two CCBs on glaucoma and analysis of their possible mechanisms Tao Liang, Lingyun Zhang, Yanhua Gao, Yanru Xiang, Yan Gao Department of Ophthalmology, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China Received April 26, 2017; Accepted May 26, 2017; Epub July 15, 2017; Published July 30, 2017 Abstract: Objective: To respectively compare the therapeutic effects of nimodipine and nifedipine on glaucoma, and then analyze the possible protective effects of these two calcium channel blockers (CCBs) on glaucomatous retinal ganglion cells (RGCs). Methods: Fifty-four patients with glaucoma were divided into control group (n=15), treat- ment group 1 (n=20) and treatment group 2 (n=19) in accordance with a random number table. General clinical treatment of glaucoma was performed in all three groups, while nimodipine was applied in treatment group 1 and nifedipine was applied in treatment group 2. The therapeutic effects and incidence of adverse reactions (intraocular pressure (IOP), eyesight, retinal light sensitivity, progressive visual field damage and adverse drug reaction) were compared among the three groups. Results: There were no significant differences in IOP and eyesight before and after treatment among the three groups (P>0.05). The retinal light sensitivity in control group began to decline from the sixth month after treatment, which was significantly different from treatment group 1 and treatment group 2 (P=0.03; P=0.04). The survival curve of visual field damage indicated that the visual field damage in control group was obviously more serious than that in the two treatment groups with the increase of sick time (P=0.03). -

Cyproheptadine Versus Propranolol in the Prevention of Migraine
Original Article Cyproheptadine versus propranolol in the prevention of migraine headaches in children Bahador Asadi1, Fariborz Khorvash2, Abolfazl Najaran3, Farzin Khorvash4 ABSTRACT Objective: There are conflicting results on the efficacy of propranolol and cyproheptadine in the prevention of migraine headaches in children. Therefore, in this study, we evaluated the efficacy of propranolol versus cyproheptadine in the prevention of migraine headaches. Methodology: This was a randomized, double-blind trial. Sixty children aged 8-15 yrs with migraine headaches were randomized to be treated with either propranolol (40-80mg per day) or cyproheptadine (8-12mg per day) for 4 weeks. The patients were requested to record the severity and duration of their headaches during a 2-week period before starting the intervention. The patients were followed at 2-week intervals for a period of 1 month after starting treatment. The headache diary was analyzed for each patient and was compared with baseline using SPSS software and statistical tests including the student’s t-test. Results: Out of 60 patients at baseline, nine patients in the cyproheptadine group and six patients in the propranolol group did not appear at the appropriate time for follow-up visits and therefore were excluded from the study. The mean age in the cyproheptadine group was 11.9 ± 2.23 years and in the propranolol group was 10.7 ± 2.33 years. Based on the diaries, the results showed that propranolol and cyproheptadine decreased headaches by 54.61% and 70.53% (p < 0.05), respectively, at the end of four weeks of treatment. Conclusion: Overall, the results of our study suggest that cyproheptadine is a good choice for prevention of migraine headache in pediatric group although more prolonged study with higher number of the patient is recommended. -

Ep 0932416 B1
Europäisches Patentamt *EP000932416B1* (19) European Patent Office Office européen des brevets (11) EP 0 932 416 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: A61K 45/06, A61K 31/485, of the grant of the patent: A61K 31/165, A61K 31/135, 22.06.2005 Bulletin 2005/25 A61K 31/00, A61K 31/275 (21) Application number: 97910772.9 (86) International application number: PCT/US1997/017828 (22) Date of filing: 06.10.1997 (87) International publication number: WO 1998/015275 (16.04.1998 Gazette 1998/15) (54) METHOD AND POTENTIATED COMPOSITION FOR TREATING MIGRAINE VERFAHREN UND POTENZIERTE ZUSAMMENSETZUNG ZUR BEHANDLUNG VON MIGRÄNE THERAPIE ET COMPOSITION PHARMACEUTIQUE POTENTIALISEE EFFICACES CONTRE LA MIGRAINE (84) Designated Contracting States: • DHAVARE ET AL: "Effect of Drugs Influencing AT BE CH DE DK ES FI FR GB GR IE IT LI LU MC Central 5-HT Mechanisms on NL PT SE Amantadine-Induced Stereotyped Behaviour in the Rat" INDIAN JOURNAL PHYSIOL. (30) Priority: 09.10.1996 US 727923 PHARMACOL., vol. 27, no. 1, 1983, pages 19-24, 24.10.1996 US 736370 XP002058885 • MAJ T ET AL: "Antagonistic Effect of (43) Date of publication of application: Cyproheptadine on Neuroleptic-Induced 04.08.1999 Bulletin 1999/31 Catalepsy" PHARMACOLOGY BIOCHEMISTRY AND BEHAVIOR, vol. 3, no. 1, 1975, pages 25-27, (73) Proprietor: Algos Pharmaceutical Corporation XP002058886 Neptune, NJ 07753 (US) • SKUZA ET AL: "Memantine, Amantadine and L-Deprenyl Potentiate the Action of L-DOPA in (72) Inventor: CARUSO, Frank, S. Monoamine-Depleted Rats" JOURNAL OF Colts Neck, NJ 07722 (US) NEURAL TRANSMISSION, vol. -

Calcium Channel Blockers
Calcium Channel Blockers Summary In general, calcium channel blockers (CCBs) are used most often for the management of hypertension and angina. There are 2 classes of CCBs: the dihydropyridines (DHPs), which have greater selectivity for vascular smooth muscle cells than for cardiac myocytes, and the non-DHPs, which have greater selectivity for cardiac myocytes and are used for cardiac arrhythmias. The DHPs cause peripheral edema, headaches, and postural hypotension most commonly, all of which are due to the peripheral vasodilatory effects of the drugs in this class of CCBs. The non-DHPs are negative inotropes and chronotropes; they can cause bradycardia and depress AV node conduction, increasing the risk of heart failure exacerbation, bradycardia, and AV block. Clevidipine is a DHP calcium channel blocker administered via continuous IV infusion and used for rapid blood pressure reductions. All CCBs are substrates of CYP3A4, but both diltiazem and verapamil are also inhibitors of 3A4 and have an increased risk of drug interactions. Verapamil also inhibits CYP2C9, CYP2C19, and CYP1A2. Pharmacology CCBs selectively inhibit the voltage-gated L-type calcium channels on cardiac myocytes, vascular smooth muscle cells, and cells within the sinoatrial (SA) and atrioventricular (AV) nodes, preventing influx of extracellular calcium. CCBs act by either deforming the channels, inhibiting ion-control gating mechanisms, and/or interfering with the release of calcium from the major cellular calcium store, the endoplasmic reticulum. Calcium influx via these channels serves for excitation-contraction coupling and electrical discharge in the heart and vasculature. A decrease in intracellular calcium will result in inhibition of the contractile process of the myocardial smooth muscle cells, resulting in dilation of the coronary and peripheral arterial vasculature. -
PDF-Document
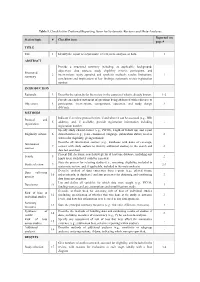
Table 1. Checklist for Preferred Reporting Items for Systematic Reviews and Meta-Analyses. Reported on Section/topic # Checklist item page # TITLE Title 1 Identify the report as a systematic review, meta-analysis, or both. 1 ABSTRACT Provide a structured summary including, as applicable: background; objectives; data sources; study eligibility criteria, participants, and Structured 2 interventions; study appraisal and synthesis methods; results; limitations; 1 summary conclusions and implications of key findings; systematic review registration number. INTRODUCTION Rationale 3 Describe the rationale for the review in the context of what is already known. 1-2 Provide an explicit statement of questions being addressed with reference to Objectives 4 participants, interventions, comparisons, outcomes, and study design 2 (PICOS). METHODS Indicate if a review protocol exists, if and where it can be accessed (e.g., Web Protocol and 5 address), and, if available, provide registration information including - registration registration number. Specify study characteristics (e.g., PICOS, length of follow-up) and report Eligibility criteria 6 characteristics (e.g., years considered, language, publication status) used as 2 criteria for eligibility, giving rationale. Describe all information sources (e.g., databases with dates of coverage, Information 7 contact with study authors to identify additional studies) in the search and 2 sources date last searched. Present full electronic search strategy for at least one database, including any Search 8 2 limits used, such that it could be repeated. State the process for selecting studies (i.e., screening, eligibility, included in Study selection 9 2-3 systematic review, and, if applicable, included in the meta-analysis). Describe method of data extraction from reports (e.g., piloted forms, Data collection 10 independently, in duplicate) and any processes for obtaining and confirming 3 process data from investigators. -

Nimodipine (Nimotop®) Information for Patients and Families
Nimodipine (Nimotop®) Information for patients and families Available as: 30 mg tablets The usual dose is 60 mg every 4 hours around the clock, for a total of 21 days. Dosing times while you were in hospital were: _____________________________________________ _____________________________________________ Remember: • You must take nimodipine at the same times every day. • Take nimodipine on an empty stomach at least 1 hour before meals or 2 hours after meals. Why am I taking this medicine? Nimodipine is used in the treatment of subarachnoid hemorrhage (bleeding in the space surrounding the brain). Nimodipine helps prevent vasospasm. Vasospasm narrows the inside of the blood vessels and reduces blood flow to the brain. Nimodipine helps keep the blood vessels open and improve blood flow to the brain. What should I do if I miss a dose? Take the dose as soon as you remember. If it is almost time for your next dose, skip the missed dose and then continue on your usual schedule. Do not take 2 doses at the same time. 2 Before you take this medicine Before you start taking nimodipine, tell your doctor if: • You are pregnant, planning on becoming pregnant or breastfeeding • You have a history heart or liver disease Where can I get this medicine? • Fill your nimodipine prescription at St. Michael’s Hospital Prescription Care Centre outpatient pharmacy. • Nimodipine is rarely used in the community, so most retail pharmacies do not stock this medicine. • Talk to your nurse, social worker, case manager or pharmacist, if you do not have drug coverage and think you may need help to cover the cost of nimodipine. -

The Effect of the Molecular Properties of Calcium Channel Blockers on Their Elimination Route
Arch. Biol. Sci., Belgrade, 67(3), 801-806, 2015 DOI:10.2298/ABS150127039T THE EFFECT OF THE MOLECULAR PROPERTIES OF CALCIUM CHANNEL BLOCKERS ON THEIR ELIMINATION ROUTE Jasna B. Trbojević-Stanković1,2, Jadranka V. Odović3, Ratomir M. Jelić4, Dejan M. Nesić5,*, Biljana B. Stojimirović1,6 1 School of Medicine, University of Belgrade, Belgrade, Serbia; 2 Department of Dialysis, Clinical Hospital Center “Dr Dragiša Mišović”, Belgrade, Serbia; 3 Department of Analytical Chemistry, Faculty of Pharmacy, University of Belgrade, Belgrade, Serbia; 4 Faculty of Medicinal Science, University of Kragujevac, Kragujevac, Serbia; 5 Institute of Medical physiology, School of Medicine, University of Belgrade, Belgrade, Serbia; 6 Clinic of Nephrology, Clinical Center of Serbia, Belgrade, Serbia; *Corresponding author: [email protected], [email protected] Abstract: Calcium channel blockers (CCBs) are among the most widely used drugs in cardiovascular medicine. In this study, nine CCBs (amlodipine, felodipine, isradipine, nicardipine, nifedipine, nimodipine, nisoldipine, verapamil and diltiazem) were investigated to assess the relationship between their molecular properties and elimination data obtained from litera- ture. The descriptors of the molecular properties of CCBs were calculated using three software packages. The relationship between computed molecular properties and elimination data collected from relevant literature, initially investigated with simple linear regression analysis, showed poor correlation (R2 <0.25). Application of molecular weight or volume data as additional independent variable, multiple linear regression (MLR) revealed better correlations (R2 ~ 0.38) between CCB renal and fecal elimination data and their lipophilicity. Excluding nimodipine from the calculations resulted in more accept- able correlations. The best correlations were established after computed lipophilicity descriptor and molecular weight were applied (R2 = 0.66 with acceptable probability value). -

A Unitary Mechanism of Calcium Antagonist Drug Action '(Dihydropyridine/Nifedipine/Verapamil/Neuroleptic/Diltiazem) KENNETH M
Proc. Nati Acad. Sci. USA Vol. 80, pp. 860-864, February 1983 Medical Sciences A unitary mechanism of calcium antagonist drug action '(dihydropyridine/nifedipine/verapamil/neuroleptic/diltiazem) KENNETH M. M. MURPHY, ROBERT J. GOULD, BRIAN L. LARGENT, AND SOLOMON H. SNYDER* Departments of Neuroscience, Pharmacology and Experimental Therapeutics, and Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, 72S North Wolfe Street, Baltimore, Maryland 21205 Contributed by Solomon H. Snyder, October 21, 1982 ABSTRACT [3H]Nitrendipine binding to drug receptor sites liquid scintillation counting were carried out as described (7). associated with calcium channels is allosterically regulated by a All experiments, performed in triplicate, were replicated at diverse group of calcium channel antagonists. Verapamil, D-600 least three times with similar results. (methoxyverapamit), tiapamil, lidoflazine, flunarizine, cinnari- Guinea pig ileum longitudinal muscles were prepared for zine, and prenylamine all reduce P3H]nitrendipine binding affin- recording as described by Rosenberger et aL (13) and incubated ity. By contrast, diltiazem, a benzothiazepine calcium channel an- in a modified Tyrode's buffer (14) at 370C with continuous aer- tagonist, enhances [3H]nitrendipine binding. All these drugeffects ation with 95% '02/5% CO2. Ileum longitudinal muscles were involve a single site allosterically linked to the [3H]nitrendipine incubated in this buffer for 30 min before Ca2"-dependent con- binding site. Inhibition of t3H]nitrendipine binding by prenyl- were as described Jim et aL amine, lidoflazine, or tiapamil is reversed by D-600and diltiazem, tractions recorded by (15). which alone respectively slightlyreduceorenhance H]mnitrendipine RESULTS binding. Diltiazem reverses the inhibition of [3H]nitrendipine binding by D-600.