Microbiological Safety Testing of Cannabis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Centrifan PE Personal Evaporator User Manual
CentrifanTM PE Personal Evaporator User Manual (Shown – CentrifanTM – PE–T; Model 78-0070 with Circulator Coil Accessory) KD Scientific 84 October Hill Road Holliston, MA 01746 Phone: (508) 429-6809 Fax: (508) 893-0160 Part 78-8425 Manual www.kdscientific.com Notices: This system is covered by a limited lifetime warranty. A copy of the warranty is included with this manual. The operator is not required to perform routine maintenance, but procedures for disassembly and cleaning are as described herein. For cleaning procedures, refer to Chapter 4. All information in this manual is subject to change without notice and does not represent a commitment on the part of KD Scientific. The system and various components in the system are the subject of various pending patents. Modular SFC was acquired by Harvard Bioscience in May 2012. KD Scientific is a division of Harvard Bioscience. No part of this manual may be reproduced or transmitted in any form or by any means without the written permission of KD Scientific. All trademarks of other companies are acknowledged to the property of the respective owners. CentrifanTM is a trademark of KD Scientific. Printed in the United States of America. Warnings and Safety Precautions The following precautions should be followed to minimize the possibility of personal injury and/or damage to property while using the CentrifanTM PE. 1. Maintain a Well-Ventilated Laboratory Volatile organic solvents may be used in the system. Ensure that the laboratory is well ventilated so that a buildup of vaporized solvent cannot occur. 2. Ensure That Solvent Vapor Is Not Allowed To Escape Into The Laboratory Any organic solvent vapor must be vented to an external venting system so that it cannot collect in the laboratory. -

Escherichia Coli HD701
The Process Intensification of Biological Hydrogen Production by Escherichia coli HD701 By Michael Sulu A thesis submitted to The University of Birmingham for the degree of DOCTOR OF PHILOSOPHY School of Chemical Engineering College of Engineering and Physical Sciences The University of Birmingham November 2009 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. Abstract Hydrogen is seen as a potential fuel for the future; its choice is driven by the increasing awareness of the necessity for clean fuel. Together with the simultaneous development of “green technologies” and sustainable development, a current goal is to convert waste to energy or to create energy from a renewable resource. Biological processing [of renewables] or bioremediation of waste to create hydrogen as a product fulfils this goal and, as such, is widely researched. In this work, an already established process, using a hydrogenase up‐regulated strain ‐ was characterised and the important process parameters were established. This bacterial strain has the potential for industrial‐scale hydrogen production from, for example, waste sugars. Previous work, repeated here, showed that hydrogen could be generated by E. -
Grout Gas Generation Test Plan ECN No
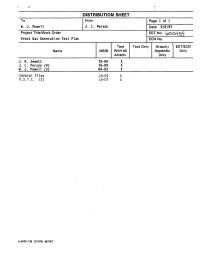
DISTRIBUTION SHEET To From Page 1 of 1 - W. J. Powell J. C. Person Date 9/8/93 Project Title/Work Order EDTNo. (pOo45^j[ Grout Gas Generation Test Plan ECN No. Text Text Only Attach./ EDT/ECN Name MSIN With All Appendix Only Attach. Only J. R. Jewett T6-09 X J. C. Person (4) T6-09 X W. J. Powell (3) R4-03 X Central Files L8-04 X 0.S.T.I. (2) L8-07 X A-6000-135 (01/93) WEF067 Page 1 of | ai p7 4QQC ENGINEERING DATA TRANSMITTAL I.EDT 600459 2. To: (Receiving Organization) 3. From: (Originating Organization) 4. Related EDT No.: Grout Technology Process Chemistry Laboratory N/A S. Proj./Prog./Dept./Div.: 6. Cog. Engr.: 7. Purchase Order No.: WM/Grout/PAL/FO J. C. Person _N/A_ 8. Originator Remarks: 9. Equip./Component No. N/A 10. Systea/Bldg./Facility: N/A 11. Receiver Remarks: 12. Major Assm. Dwg. No. 13. Permit/Permit Application No.: 14. Required Response Date: 15. DATA TRANSMITTED (F) (G) (H) (I) (A| (C) (D) Reason Origi• Receiv• (E) Title or Description of Data Impact Item Sheet Rev. for nator er (B) Document/Drawing No. Transmitted Level No. No. No. Trans• Dispo• Dispo• mittal sition sition 1. WHCSD-WM-TP-180 Grout Gas Generation 3Q Test Plan 16. KEY Impact Level (F) Reason for Transmittal (G) Disposition (H) & (I) 1, 2, 3, or 4 (see 1. Approval 4. Review 1. Approved 4. Reviewed no/comment MRP 5.431 3Q 2. Release 5. Post-Review 2. Approved w/comment 5. -

KNF Rotary Evaporator Brochure
RC 900 AND RC 600 ROTARY EVAPORATORS. INSPIRINGLY EASY TO USE. SYSTEMS EXPERTISE BENEFIT FROM EXPERT KNOWLEDGE. ROTARY EVAPORATION TAILORED TO PRACTICAL NEEDS. Under the spotlight at KNF: What aspects are really key What makes KNF’s rotary evaporators stand out? For to rotary evaporation in everyday lab practice? What is years we have offered the SC 920 vacuum pump system, needed to guarantee simple, economical and reliable a much sought-after laboratory device that works side by processes day in day out? These are the questions we side with distilling apparatus. Simple handling, a small used to guide us when developing and implementing the footprint and whisper quiet operation are the KNF RC 900 and the new RC 600. We became involved in daily strengths that set the SC 920 apart. lab work. We asked lab technicians what they wished for, enlisted experts to perform tests, and incorporated their Its success has encouraged us to broaden our systems suggestions. approach to create a perfectly balanced package comprising The result? KNF rotary evaporators designed to impress a rotary evaporator, vacuum pump system, and chiller. thanks to their distinct handling advantages, clever Curious? Then discover KNF’s rotary evaporators and functional details and well thought out safety features. their advantages today! EASY TO USE | CLEVER FUNCTIONAL DETAILS | WELL THOUGHT OUT SAFETY FEATURES RC 900. SUPERIOR PERFORMANCE SYSTEM. RC 600. DUAL EVAPORATION FOR ACADEMIA LABS. Rotary evaporator, vacuum pump system, and chiller as a perfectly System packages to suit different budget conditions are available – coordinated system. e.g., one comprising a vacuum pump system to simultaneously and independently assist two rotary evaporators. -

FEI Tecnai 12 ('T12') 120 Kv
Ver. 2017-07-07 Emergency Information: 1. Medical Emergencies: Contact 911 and McGill Security 514.398.3000 2. Leave TEM as is. Do NOT shut down the vacuum system. 3. If possible, turn off High Tension and Close Column Valve. 4. Exit the Room/Building. Emergency Contact Information: • Jeannie Mui – EM Coordinator: Office 514.398.6329; Email: [email protected] • S. Kelly Sears – Facility Manager: Cell 514.576.1926; Email: [email protected] • Joaquin Ortega – Director: Office 514.398.6348; [email protected] FEI Tecnai 12 (‘T12’) 120 kV TEM Simplified Operating Manual (Prepared by S Kelly Sears and Joaquin Ortega) Safety • Do NOT wander behind the microscope or step on cables. • Do NOT touch the sample rod with bare hands below the black O-ring. • There are two computers on the T12 TEM: 1. Microscope PC (MPC) that controls the TEM • Note: Under no circumstances should a user insert a USB device into the MPC. 2. Support PC (SPC) to store and retrieve images (on the desk to the left of the TEM). Protective Equipment • Nitrile gloves for handling sample holder and safety glasses for filling liquid nitrogen dewar. I. Microscope Start Up Note: Your PC desktop may not appear exactly as shown in the images in this manual. a. VERIFY the Red On/Off button and the White Vac. and HT buttons on the right console are lit (currently NOT WORKING). b. LOG IN to the MPC with the username and password created during your training session. After logging in, VERIFY the microscope icon on the right side of the bottom task bar is present and green. -

INNOVATIVE BIOPROCESS EQUIPMENT 2 SOLIDABIOTECH.Com 3
SOLIDABIOTECH.coM INNOVATIVE BIOPROCESS EQUIPMENT 2 SOLIDABIOTECH.coM 3 Solida Biotech is the ideal bioprocess partner to find the solution that suits your specific needs SOLIDA BIOTECH We are a dynamic company that specialises in engineering, developing, and manufacturing in- novative bioprocess equipment. The company was founded by a management team with over 20 years’ experience in the fields of fermentation, cell culture, sterile processing, software and automation technologies, with the aim of meeting the challenges of producing and supplying leading industrial and institutional customers with a new generation of bioreactors and fermenters for research and production, as well as scientific instruments. THE IDEAL BIOPROCESS partner Thanks to the abilities of our skilled team, we can offer flexible and custom made solutions to deal with multiple requirements in the world of green, red and white biotechnologies. Solida offers a wide range of compact and modular bioreactors and fermenters from 50ml to 50 cubic metre pilot and industrial plants and integrated turn-key solutions. All our equipment meets the standards for worldwide distribution. bioprocess solutions We manufacture a wide range of bioreactors, fermenters, at-line analysers, CIP/SIP systems and process vessels for research, development, and production, suitable for fermentation, cell culture, stem cells, re- newable technologies, algae, biomass, biofuel, hydrolysis and more. coMpact bioreactors LaboratorY SIP Bioreactors A compact laboratory bioreactor solution Modular Laboratory SIP stainless steel in characterised by great performance and ease of situ sterilisable bioreactors, from 1 litre up to 50 use, the perfect way to get started. litre w/v as well as custom fit solutions. -

SOP Diazomethane.Pdf
STANDARD OPERATING PROCEDURE: DIAZOMETHANE GENERATION1 PI: Hans-Joachim Lehmler Room & Building: 223 IREH Department: OEH Research Group: Lehmler/Ludewig/Robertson Date: 03/22/2009 DESCRIPTION Diazomethane is a common methylating reagent for carboxylic acids. It has found wide application in the methylation of phenols, enols (alkenols), and heteroatoms such as nitrogen and sulfur. PROCEDURE Please see the attached experimental protocol for the generation of diazomethane. MATERIALS & HAZARDS Influe nced by the scale of production, several methods are available for the generation of diazomethane, each requiring a different suite of materials/chemicals. ic Sensitive Reactive - Principal Materials Used - (CAS No.) Other Comments: Irritant Sensitizer toxin Reproductive Tox Acutely Carcinogen Flammable Combustible Water Shock Pyrophoric Oxidizer Biotoxin Corrosive Diazomethane (334-88-3) X X X X X See comment 1, below. Diazald (85-11-5) X X Ethanol (64-17-5) X X X Potassium hydroxide (1310-58-3) X X See comment 3, below. Sodium hydroxide (1310-73-2) X X See comment 3, below. Acetone (67-64-1) X X Ether (60-29-7) X X X X See comment 4, below. Other comments: 1. Diazomethane is extremely toxic and highly irritating, causing pulmonary edema when inhaled in high concentrations. Long-term, low-level exposure may lead to sensitization, resulting in asthma-like symptoms. Diazomethane has also been known to explode unaccountably, both as a gas and in solution. Rough surfaces are proven initiators of detonation. - 1 - 2. Potassium hydroxide contact with moisture or water may generate sufficient heat to ignite combustible materials. 3. E ther is extremely flammable. It forms explosive peroxides after prolonged exposure to light and air. -

Unique PTI Quantamaster™ Benefits
TM PTI QuantaMaster Series Modular Research Fluorometers for Lifetime and Steady State Measurements Most sensitive and flexible fluorometers www.quantamaster.com Unique PTI QuantaMaster™ Benefits • Highest guaranteed sensitivity specification • World class software for all steady state and lifetimes needs • Extended wavelength range with triple grating monochromators • Multiple light sources and detectors with dual entrance and dual exit monochromators • Excellent stray light rejection with large single or double additive, coma corrected, monochromators • World class TCSPC lifetime enhancements, up to 100 MHz • NIR steady state and phosphorescence lifetime detection to 5,500 nm “The new PTI QuantaMaster 8000 series of modular research fluorometers from HORIBA Scientific offer the world’s highest guaranteed sensitivity specifications, plus many unique benefits.” PTI QuantaMaster 8000 Series The PTI QuantaMaster™ series of modular research grade spectrofluorometers are multidimensional systems for photoluminescence measurements. The foundation of a fluorescence spectroscopy laboratory is built on steady state intensity measurements such as wavelength scans, time-based experiments, synchronous scans and polarization. The PTI QuantaMaster 8000 series ensures you get the best possible results for all these measurements with high sensitivity, spectral resolution and stray light rejection. This level of sensitivity is achieved using a unique xenon illuminator, providing safety, cost, and energy consumption benefits not found amongst competitor companies. These conditions make the PTI QuantaMaster system ideal for the highest demands of fluorescence research. The PTI QuantaMaster system is adaptable to every research need, with additions such as TCSPC fluorescence lifetimes, upconversion lasers and phosphorescence detection up to 5,500 nm. Using an optional pulsed light source allows for not only spectral and kinetic fluorescence and phosphorescence measurements, but also the measurement of lifetimes in the picosecond to seconds range. -

Organic Chemistry Laboratory Experiments for Organic Chemistry Laboratory 860-121-02 Mw 1:00-4:00
ORGANIC CHEMISTRY LABORATORY EXPERIMENTS FOR ORGANIC CHEMISTRY LABORATORY 860-121-02 MW 1:00-4:00 WRITTEN, COMPILED AND EDITED BY LINDA PAAR JEFFREY ELBERT KIRK MANFREDI SPRING 2008 TABLE OF CONTENTS SYNTHESIS OF ASPIRIN 1 MELTING POINT AND CRYSTALLIZATION 2 DISTILLATION 8 EXTRACTION 11 TLC AND CHROMATOGRAPHY 14 NATURAL PRODUCTS: ISOLATION OF LIMONENE 23 FREE RADICAL CHLORINATION 24 SN1 AND SN2 REACTIONS 27 DEHYDRATION REACTIONS 30 GRIGNARD SYNTHESIS 32 COMPUTATIONAL CHEMISTRY 36 MULTIPLE STEP SYNTHESIS 38 ORGANIC CHEMISTRY 121 EXPERIMENT 1 SYNTHESIS OF ASPIRIN FROM SALICYLIC ACID Aspirin is one of the oldest and most common drugs in use today. It is both an analgesic (pain killer) and antipyretic (reduces fever). One method of preparation is to react salicylic acid (1 ) with acetic anhydride (2) and a trace amount of acid (equation 1). O OH CH3 O COOH COOH + H + CH3COOH + (CH3CO)2O 3 4 1 2 The chemical name for aspirin is acetylsalicylic acid (3) PROCEDURE Place 3.00 g of salicylic acid in a 125 ml Erlenmeyer flask. Cautiously add 6 ml of acetic anhydride and then 5 drops of concentrated H2SO4. Mix the reagents and heat the flask in a beaker of water warmed to 80-90°C, for 10 minutes. Remove the Erlenmeyer flask and allow it to cool to room temperature. Add 40 ml of H2O and let the sample crystallize in an ice-water bath.* Filter and wash the crystals with cold water. Allow them to air dry overnight and weigh the product. What is the percent yield? One drawback to this synthetic procedure is that there is the possibility of some left over salicylic acid. -

Rotary Evaporators
Rotary Evaporators Rotary evaporators are commonly used for separating solvents, the Stuart ® range offers simple control and a variety of glassware configurations, vertical, diagonal and cold finger condensing depending on your requirements. All configurations are available with plastic coated glass. Page 76 - Rotary evaporators Page 78 - Glassware accessories and spares Page 79 - Water bath Page 80 - Vacuum Pump - Recirculating cooler THE BENCHTOP EQUIPMENT HYGIENE SOLUTION Stuart Science Equipment Catalogue Page 75 Rotary Evaporators Rotary evaporators • Simple, counterbalanced lift mechanism • PTFE/glass liquid pathway for chemical inertness • Sparkless induction motor • Long life graphite impregnated PTFE vacuum seal • Efficient flask and vapour tube ejection system Rotary evaporators are distillation units that incorporate an efficient condenser with a rotary flask system. As the flask containing the solvent is rotated it continually transfers a thin layer of liquid over the entire inner surface. This gives a very large surface area for evaporation that is effected by heating from the accessory waterbath (see page 79). They are the ideal tools for many everyday laboratory applications including:- • Concentration of solutions • Reclamation of solvents • Vacuum drying of wet solids • Degassing liquids The rotating system is fitted with a special seal that allows the apparatus to be placed under vacuum. This reduces the boiling point of the solvents and removes the vapour phase making the process much more efficient. See page 80 for details of the vacuum pump. Each unit is also provided with an easy to use vacuum release and a continuous feed system, which allows more solvent to be drawn into the rotating Florentine flask without the need to stop the operation. -

Application of Azeotropic Distillation to the Separation of Olefins And
Application of azeotropic distillation to the separation of olefins and paraffins by Padmanabh D Datar A thesis submitted to the Graduate Faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Chemical Engineering Montana State University © Copyright by Padmanabh D Datar (1966) Abstract: The purpose of this investigation was to study the possible application of azeotropic distillation to the separation pf olefins and paraffins. An attempt was made to enhance the ease of separation of two pairs of compounds by using azeotropic distillation. The two pairs were 2„2,4 - trimethyl pentane, 2,4.,4 - trimethyl pentene-1, and normal heptane - 2,4,4 - trimethyl pentene-1. The degree to which a compound (called entrainer) helped separation was measured by the increase in the relative volatility of the particular pair of compounds being separated over the value obtained by distilling the pure compounds without entrainer. Compounds picked as entrainers were those which contained oxygen, nitrogen or halogen atoms and were for these reasons likely to give large deviations from Raoult's Law in solution. The compounds were picked to boil within about ± 20°C of the hydrocarbons being separated. Twenty-two compounds satisfying these requirements were tried as possible entrainers. Major pieces of equipment used included four identical distillation columns packed with Fenske rings and fitted with cold finger condensing head, a gas chromatograph, and a refractometer. The evaluation of an entrainer consisted of determining whether or not an azeotrope was formed, determining the azeotropic composition, and determining the relative volatility between the hydrocarbons using the entrainer. -
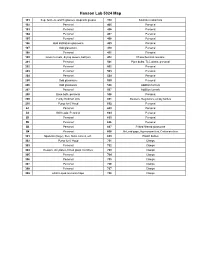
Hanson Group Lab Maps
Hanson Lab 5024 Map 101 Sep. funnels, watch glasses, stopcock grease 310 Soxholet extractors 102 Personal 405 Personal 103 Personal 406 Personal 104 Personal 407 Personal 105 Personal 408 Personal 106 Odd distillation glassware 409 Personal 107 Odd glassware 410 Personal 108 Personal 411 Personal 109 Glass funnels, drying towers, bell jars 412 Photochemical reactors 201 Personal 501 Pipet bulbs, TLC plates, personal 202 Personal 502 Personal 203 Personal 503 Personal 204 Personal 504 Personal 205 Odd glassware 505 Personal 206 Odd glassware 506 Addition funnels 207 Personal 507 Addition funnels 208 Base bath, personal 508 Personal 209 Funky Hickman stills 601 Beakers, Regulators, empty bottles 210 Pump for 6' Hood 602 Personal A1 Personal 603 Personal A2 Still heads, Personal 604 Personal B1 Personal 605 Personal B2 Personal 606 Personal B3 Personal 607 Fritted filtered glassware B4 Personal 608 McLeod gage, Hg monameters, Cartesian diver 301 Spatulas (large), files, forks, knives, etc. 609 Plastic bottles 302 Pump for 6' Hood 701 Clamps 303 Personal 702 Clamps 304 Dewars, stir plates, fritted glass crucibles 703 Clamps 305 Personal 704 Clamps 306 Personal 705 Clamps 307 Personal 706 Clamps 308 Personal 707 Clamps 309 24/40 Liquid ammonia traps 708 Clamps 102 104 Door to Sink Givens Solvent 103 105 Lab 101 109 Desk Cabinet 106 108 107 N Door to Hallway 206 A2 A1 210 208 205 207 Inorganic 204 202 6' Hood Sink Acids 209 203 201 305 307 6' Hood Sink 301 306 308 304 B1 B3 309 302 303 B2 B4 310 412 405 411 406 8' Hood 410 408 8' Sink Hood