Genome-Wide Next-Generation DNA and RNA Sequencing Reveals A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

High Mutation Frequency of the PIGA Gene in T Cells Results In
High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI A nchor−/CD52− T Cells That Can Give Early Immune Protection after This information is current as Alemtuzumab-Based T Cell−Depleted of October 1, 2021. Allogeneic Stem Cell Transplantation Floris C. Loeff, J. H. Frederik Falkenburg, Lois Hageman, Wesley Huisman, Sabrina A. J. Veld, H. M. Esther van Egmond, Marian van de Meent, Peter A. von dem Borne, Hendrik Veelken, Constantijn J. M. Halkes and Inge Jedema Downloaded from J Immunol published online 2 February 2018 http://www.jimmunol.org/content/early/2018/02/02/jimmun ol.1701018 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2018/02/02/jimmunol.170101 Material 8.DCSupplemental Why The JI? Submit online. by guest on October 1, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published February 2, 2018, doi:10.4049/jimmunol.1701018 The Journal of Immunology High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI Anchor2/CD522 T Cells That Can Give Early Immune Protection after Alemtuzumab- Based T Cell–Depleted Allogeneic Stem Cell Transplantation Floris C. -

Biosynthesis and Deficiencies of Glycosylphosphatidylinositol
130 Proc. Jpn. Acad., Ser. B 90 (2014) [Vol. 90, Review Biosynthesis and deficiencies of glycosylphosphatidylinositol † By Taroh KINOSHITA*1, (Communicated by Kunihiko SUZUKI, M.J.A.) Abstract: At least 150 different human proteins are anchored to the outer leaflet of the plasma membrane via glycosylphosphatidylinositol (GPI). GPI preassembled in the endoplasmic reticulum is attached to the protein’s carboxyl-terminus as a post-translational modification by GPI transamidase. Twenty-two PIG (for Phosphatidyl Inositol Glycan) genes are involved in the biosynthesis and protein-attachment of GPI. After attachment to proteins, both lipid and glycan moieties of GPI are structurally remodeled in the endoplasmic reticulum and Golgi apparatus. Four PGAP (for Post GPI Attachment to Proteins) genes are involved in the remodeling of GPI. GPI- anchor deficiencies caused by somatic and germline mutations in the PIG and PGAP genes have been found and characterized. The characteristics of the 26 PIG and PGAP genes and the GPI deficiencies caused by mutations in these genes are reviewed. Keywords: glycosylphosphatidylinositol, glycolipid, post-translational modification, somatic mutation, germline mutation, deficiency glycolipid membrane anchors. GPI-APs are typical Introduction raft-associated proteins and tend to form homo- At least 150 different human proteins are post- dimers.3),4) GPI-APs can be released from the translationally modified by glycosylphosphatidylino- cell after cleavage by GPI-cleaving enzymes or sitol (GPI) at the carboxyl (C)-terminus.1),2) These GPIases.5)–8) These characteristics are critical for proteins are expressed on the cell surface by being the functions of individual GPI-APs and for embryo- anchored to the outer leaflet of the plasma membrane genesis, neurogenesis, fertilization, and the immune via the phosphatidylinositol (PI) moiety and termed system.5),8)–11) GPI-anchored proteins (GPI-APs). -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Genetic Tools to Improve Reproduction Traits in Dairy Cattle
Genetic tools to improve reproduction traits in dairy cattle Aurelien Capitan, Pauline Michot, Aurélia Baur, Romain Saintilan, Chris Hoze, Damien Valour, François Guillaume, D Boichon, Anne Barbat, Didier Boichard, et al. To cite this version: Aurelien Capitan, Pauline Michot, Aurélia Baur, Romain Saintilan, Chris Hoze, et al.. Genetic tools to improve reproduction traits in dairy cattle. Reproduction, Fertility and Development, CSIRO Publishing, 2015, 27 (1), pp.14-21. 10.1071/RD14379. hal-01194016 HAL Id: hal-01194016 https://hal.archives-ouvertes.fr/hal-01194016 Submitted on 28 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. CSIRO PUBLISHING Reproduction, Fertility and Development, 2015, 27, 14–21 http://dx.doi.org/10.1071/RD14379 Genetic tools to improve reproduction traits in dairy cattle A. CapitanA,B,F, P. MichotA,B, A. BaurA,B, R. SaintilanA,B, C. Hoze´ A,B, D. ValourA,D, F. GuillaumeC, D. BoichonE, A. BarbatB, D. BoichardB, L. SchiblerA and S. FritzA,B AUNCEIA (Union Nationale des Coope´ratives d’Elevage et d’Inse´mination Animale), 149 rue de Bercy, 75012 Paris, France. BINRA (Institut National de la Recherche Agronomique), UMR1313 Ge´ne´tique Animale et Biologie Inte´grative, Domaine de Vilvert, 78352 Jouy-en-Josas, France. -

Runs of Homozygosity in Sub-Saharan African Populations
Human Genetics (2019) 138:1123–1142 https://doi.org/10.1007/s00439-019-02045-1 ORIGINAL INVESTIGATION Runs of homozygosity in sub‑Saharan African populations provide insights into complex demographic histories Francisco C. Ceballos1 · Scott Hazelhurst1,2 · Michèle Ramsay1,3 Received: 8 April 2019 / Accepted: 3 July 2019 / Published online: 16 July 2019 © Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract The study of runs of homozygosity (ROH) can shed light on population demographic history and cultural practices. We present a fne-scale ROH analysis of 1679 individuals from 28 sub-Saharan African (SSA) populations along with 1384 individuals from 17 worldwide populations. Using high-density SNP coverage, we could accurately identify ROH > 300 kb using PLINK software. The genomic distribution of ROH was analysed through the identifcation of ROH islands and regions of heterozygosity (RHZ). The analyses showed a heterogeneous distribution of autozygosity across SSA, revealing complex demographic histories. They highlight diferences between African groups and can diferentiate the impact of consanguine- ous practices (e.g. among the Somali) from endogamy (e.g. among several Khoe and San groups). Homozygosity cold and hotspots were shown to harbour multiple protein coding genes. Studying ROH therefore not only sheds light on population history, but can also be used to study genetic variation related to adaptation and potentially to the health of extant populations. Introduction of African populations, a study on runs of homozygosity (ROH) provides an interesting opportunity for a deep dive African human genetic diversity provides the ideal backdrop into the demographic history of Africans. to reconstruct modern human origins, the genetic basis of ROH are contiguous regions of the genome where an indi- adaptation to diferent environments and the development vidual is homozygous (autozygous) across all sites (Cebal- of more efective vaccines (Campbell and Tishkof 2008). -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -
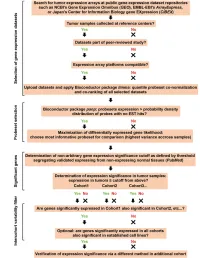
Supplementary Data
SUPPLEMENTAL INFORMATION A study restricted to chemokine receptors as well as a genome-wide transcript analysis uncovered CXCR4 as preferentially expressed in Ewing's sarcoma (Ewing's sarcoma) cells of metastatic origin (Figure 4). Transcriptome analyses showed that in addition to CXCR4, genes known to support cell motility and invasion topped the list of genes preferentially expressed in metastasis-derived cells (Figure 4D). These included kynurenine 3-monooxygenase (KMO), galectin-1 (LGALS1), gastrin-releasing peptide (GRP), procollagen C-endopeptidase enhancer (PCOLCE), and ephrin receptor B (EPHB3). KMO, a key enzyme of tryptophan catabolism, has not been linked to metastasis. Tryptophan and its catabolites, however, are involved in immune evasion by tumors, a process that can assist in tumor progression and metastasis (1). LGALS1, GRP, PCOLCE and EPHB3 have been linked to tumor progression and metastasis of several cancers (2-4). Top genes preferentially expressed in L-EDCL included genes that suppress cell motility and/or potentiate cell adhesion such as plakophilin 1 (PKP1), neuropeptide Y (NPY), or the metastasis suppressor TXNIP (5-7) (Figure 4D). Overall, L-EDCL were enriched in gene sets geared at optimizing nutrient transport and usage (Figure 4D; Supplementary Table 3), a state that may support the early stages of tumor growth. Once tumor growth outpaces nutrient and oxygen supplies, gene expression programs are usually switched to hypoxic response and neoangiogenesis, which ultimately lead to tumor egress and metastasis. Accordingly, gene sets involved in extracellular matrix remodeling, MAPK signaling, and response to hypoxia were up-regulated in M-EDCL (Figure 4D; Supplementary Table 4), consistent with their association to metastasis in other cancers (8, 9). -
Chromosome 14 Deletions, Rings, and Epilepsy Genes: a Riddle Wrapped in a Mystery Inside an Enigma
Received: 24 June 2020 | Revised: 16 October 2020 | Accepted: 16 October 2020 DOI: 10.1111/epi.16754 CRITICAL REVIEW – INVITED COMMENTARY Chromosome 14 deletions, rings, and epilepsy genes: A riddle wrapped in a mystery inside an enigma Alessandro Vaisfeld1 | Serena Spartano1 | Giuseppe Gobbi2 | Annamaria Vezzani3 | Giovanni Neri1,4 1Institute of Genomic Medicine, Catholic University School of Medicine, Rome, Italy Abstract 2Residential Center for Rehabilitation Luce The ring 14 syndrome is a rare condition caused by the rearrangement of one chromo- Sul Mare, Rimini, Italy some 14 into a ring-like structure. The formation of the ring requires two breakpoints 3 Department of Neuroscience, IRCCS- and loss of material from the short and long arms of the chromosome. Like many Istituto di Ricerche Farmacologiche Mario other chromosome syndromes, it is characterized by multiple congenital anomalies Negri, Milano, Italy 4J.C. Self Research Institute, Greenwood and developmental delays. Typical of the condition are retinal anomalies and drug- Genetic Center, Greenwood, SC, USA resistant epilepsy. These latter manifestations are not found in individuals who are carriers of comparable 14q deletions without formation of a ring (linear deletions). Correspondence Giovanni Neri, Institute of Genomic To find an explanation for this apparent discrepancy and gain insight into the mecha- Medicine, Catholic University School of nisms leading to seizures, we reviewed and compared literature cases of both ring Medicine, Rome, Italy. and linear deletion syndrome with respect to both their clinical manifestations and Email: [email protected] the role and function of potentially epileptogenic genes. Knowledge of the epilepsy- Funding information related genes in chromosome 14 is an important premise for the search of new and Ring 14 International; Ring14 Italia effective drugs to combat seizures. -

The Glycosylphosphatidylinositol Biosynthesis Pathway in Human Diseases Tenghui Wu1,2, Fei Yin1,2, Shiqi Guang1,2, Fang He1,2, Li Yang1,2 and Jing Peng1,2*
Wu et al. Orphanet Journal of Rare Diseases (2020) 15:129 https://doi.org/10.1186/s13023-020-01401-z REVIEW Open Access The Glycosylphosphatidylinositol biosynthesis pathway in human diseases Tenghui Wu1,2, Fei Yin1,2, Shiqi Guang1,2, Fang He1,2, Li Yang1,2 and Jing Peng1,2* Abstract Glycosylphosphatidylinositol biosynthesis defects cause rare genetic disorders characterised by developmental delay/ intellectual disability, seizures, dysmorphic features, and diverse congenital anomalies associated with a wide range of additional features (hypotonia, hearing loss, elevated alkaline phosphatase, and several other features). Glycosylphosphatidylinositol functions as an anchor to link cell membranes and protein. These proteins function as enzymes, adhesion molecules, complement regulators, or co-receptors in signal transduction pathways. Biallelic variants involved in the glycosylphosphatidylinositol anchored proteins biosynthetic pathway are responsible for a growing number of disorders, including multiple congenital anomalies-hypotonia-seizures syndrome; hyperphosphatasia with mental retardation syndrome/Mabry syndrome; coloboma, congenital heart disease, ichthyosiform dermatosis, mental retardation, and ear anomalies/epilepsy syndrome; and early infantile epileptic encephalopathy-55. This review focuses on the current understanding of Glycosylphosphatidylinositol biosynthesis defects and the associated genes to further understand its wide phenotype spectrum. Keywords: GPI-APs, PIG/PGAP genes, Phenotype Introduction can be divided into the