The Impact of Genetic Diversity on Gene Essentiality Within the E. Coli Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Rnase H-Like Superfamily: New Members, Comparative Structural Analysis and Evolutionary Classification Karolina A
4160–4179 Nucleic Acids Research, 2014, Vol. 42, No. 7 Published online 23 January 2014 doi:10.1093/nar/gkt1414 The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification Karolina A. Majorek1,2,3,y, Stanislaw Dunin-Horkawicz1,y, Kamil Steczkiewicz4, Anna Muszewska4,5, Marcin Nowotny6, Krzysztof Ginalski4 and Janusz M. Bujnicki1,3,* 1Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology, ul. Ks. Trojdena 4, PL-02-109 Warsaw, Poland, 2Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA USA-22908, USA, 3Bioinformatics Laboratory, Institute of Molecular Biology and Biotechnology, Adam Mickiewicz University, Umultowska 89, PL-61-614 Poznan, Poland, 4Laboratory of Bioinformatics and Systems Biology, Centre of New Technologies, University of Warsaw, Zwirki i Wigury 93, PL-02-089 Warsaw, Poland, 5Institute of Biochemistry and Biophysics PAS, Pawinskiego 5A, PL-02-106 Warsaw, Poland and 6Laboratory of Protein Structure, International Institute of Molecular and Cell Biology, ul. Ks. Trojdena 4, PL-02-109 Warsaw, Poland Received September 23, 2013; Revised December 12, 2013; Accepted December 26, 2013 ABSTRACT revealed a correlation between the orientation of Ribonuclease H-like (RNHL) superfamily, also called the C-terminal helix with the exonuclease/endo- the retroviral integrase superfamily, groups together nuclease function and the architecture of the numerous enzymes involved in nucleic acid metab- active site. Our analysis provides a comprehensive olism and implicated in many biological processes, picture of sequence-structure-function relation- including replication, homologous recombination, ships in the RNHL superfamily that may guide func- DNA repair, transposition and RNA interference. -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Anamika Kothari Quang Ong Ron Caspi An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Peter D Karp Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Csac1394711Cyc: Candidatus Saccharibacteria bacterium RAAC3_TM7_1 Cellular Overview Connections between pathways are omitted for legibility. Tim Holland TM7C00001G0420 TM7C00001G0109 TM7C00001G0953 TM7C00001G0666 TM7C00001G0203 TM7C00001G0886 TM7C00001G0113 TM7C00001G0247 TM7C00001G0735 TM7C00001G0001 TM7C00001G0509 TM7C00001G0264 TM7C00001G0176 TM7C00001G0342 TM7C00001G0055 TM7C00001G0120 TM7C00001G0642 TM7C00001G0837 TM7C00001G0101 TM7C00001G0559 TM7C00001G0810 TM7C00001G0656 TM7C00001G0180 TM7C00001G0742 TM7C00001G0128 TM7C00001G0831 TM7C00001G0517 TM7C00001G0238 TM7C00001G0079 TM7C00001G0111 TM7C00001G0961 TM7C00001G0743 TM7C00001G0893 TM7C00001G0630 TM7C00001G0360 TM7C00001G0616 TM7C00001G0162 TM7C00001G0006 TM7C00001G0365 TM7C00001G0596 TM7C00001G0141 TM7C00001G0689 TM7C00001G0273 TM7C00001G0126 TM7C00001G0717 TM7C00001G0110 TM7C00001G0278 TM7C00001G0734 TM7C00001G0444 TM7C00001G0019 TM7C00001G0381 TM7C00001G0874 TM7C00001G0318 TM7C00001G0451 TM7C00001G0306 TM7C00001G0928 TM7C00001G0622 TM7C00001G0150 TM7C00001G0439 TM7C00001G0233 TM7C00001G0462 TM7C00001G0421 TM7C00001G0220 TM7C00001G0276 TM7C00001G0054 TM7C00001G0419 TM7C00001G0252 TM7C00001G0592 TM7C00001G0628 TM7C00001G0200 TM7C00001G0709 TM7C00001G0025 TM7C00001G0846 TM7C00001G0163 TM7C00001G0142 TM7C00001G0895 TM7C00001G0930 Detoxification Carbohydrate Biosynthesis DNA combined with a 2'- di-trans,octa-cis a 2'- Amino Acid Degradation an L-methionyl- TM7C00001G0190 superpathway of pyrimidine deoxyribonucleotides de novo biosynthesis (E. -

Prediction of Novel Inhibitors Against Exodeoxyribonuclease І of H
International Journal of Scientific & Engineering Research, Volume 6, Issue 2, February-2015 217 ISSN 2229-5518 Prediction of Novel Inhibitors against Exodeoxyribonuclease І of H. influenzae through In Silico Approach Shaik Parveen, Natarajan Pradeep, Kanipakam Hema and Amineni Umamaheswari Abstract — These Haemophilus influenzae is a Gram-negative bacterium which causes pneumonia in humans. Due to its multidrug resistance to peni- cillin, rifampin and polymyxin and adverse effects of existing treatments, the condition became an open challenge for many researchers to discover novel antagonists for the treatment of pneumonia caused by H. influenzae. 5 potential sRNA candidates were identified in H. influenzae using sRNA predict tool, among them3 were enzymes and 2 were non-enzymes. Among the three identified enzymes exodeoxyribonuclease І of H. influenzae was non- homologous to humans was selected as novel drug target in the present study. Exodeoxyribonuclease І is an enzyme involved in the mismatch repair mechanism of H. influenzae. The 3D structure of the exodeoxyribonuclease І was modeled based on the crystal structure of 4JRP using Modeler 9v13 and built model was validated using PROCHECK analysis, ProQ and ProSA. The existing eight inhibitors of exodeoxyribonuclease І were searched in 3D ligand database through shape screening against ASINEX database using Phasev3.2 module and structural analogs were docked with exodeoxyri- bonuclease І in Maestro v9.6 virtual screening workflow, that implements three stage Glide docking protocol. The docking results revealed that 11 leads were having better docking and ∆G scores when compared with the eight existing inhibitors among them lead 1 was having ∆G score of -68.78 kcal/mol. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0058468 A1 Mickeown (43) Pub
US 20120058468A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0058468 A1 MickeoWn (43) Pub. Date: Mar. 8, 2012 (54) ADAPTORS FOR NUCLECACID Related U.S. Application Data CONSTRUCTS IN TRANSMEMBRANE (60) Provisional application No. 61/148.737, filed on Jan. SEQUENCING 30, 2009. Publication Classification (75) Inventor: Brian Mckeown, Oxon (GB) (51) Int. Cl. CI2O I/68 (2006.01) (73) Assignee: OXFORD NANOPORE C7H 2L/00 (2006.01) TECHNOLGIES LIMITED, (52) U.S. Cl. ......................................... 435/6.1:536/23.1 Oxford (GB) (57) ABSTRACT (21) Appl. No.: 13/147,159 The invention relates to adaptors for sequencing nucleic acids. The adaptors may be used to generate single stranded constructs of nucleic acid for sequencing purposes. Such (22) PCT Fled: Jan. 29, 2010 constructs may contain both strands from a double stranded deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) (86) PCT NO.: PCT/GB1O/OO160 template. The invention also relates to the constructs gener ated using the adaptors, methods of making the adaptors and S371 (c)(1), constructs, as well as methods of sequencing double stranded (2), (4) Date: Nov. 15, 2011 nucleic acids. Patent Application Publication Mar. 8, 2012 Sheet 1 of 4 US 2012/0058468 A1 Figure 5' 3 Figure 2 Figare 3 Patent Application Publication Mar. 8, 2012 Sheet 2 of 4 US 2012/0058468 A1 Figure 4 End repair Acid adapters ligate adapters Patent Application Publication Mar. 8, 2012 Sheet 3 of 4 US 2012/0058468 A1 Figure 5 Wash away type ifype foducts irrirrosilise Type| capture WashRE products away unbcure Pace É: Wash away unbound rag terts Free told fasgirre?ts aid raiser to festible Figure 6 Beatre Patent Application Publication Mar. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Conservation and Diversity of Radiation and Oxidative Stress Resistance
FEMS Microbiology Reviews, fuy037, 43, 2019, 19–52 doi: 10.1093/femsre/fuy037 Advance Access Publication Date: 18 October 2018 Review Article REVIEW ARTICLE Conservation and diversity of radiation and oxidative stress resistance mechanisms in Deinococcus species Sangyong Lim1, Jong-Hyun Jung1, Laurence Blanchard2 † and Arjan de Groot2,∗, 1Biotechnology Research Division, Korea Atomic Energy Research Institute, Jeongeup 56212, Republic of Korea and 2Aix Marseille Univ, CEA, CNRS, BIAM, Saint Paul-Lez-Durance, France ∗Corresponding author: DRF/BIAM/LBC, UMR 7265 CEA-CNRS-AMU CEA, Cadarache, 13108 Saint Paul Lez Durance, France. Tel: +33 04 42 25 39 35; Fax: +33 04 42 25 47 01; E-mail: [email protected] One sentence summary: The authors reviewed the mechanisms and factors involved in the extreme radiation and oxidative stress resistance in Deinococcus radiodurans in comparison with 10 other resistant Deinococcus species, and highlighted not only conserved pathways but also a large diversity of the repair, protection and regulation toolbox among the different deinococci. Editor: Kenn Gerdes †Arjan de Groot, http://orcid.org/0000-0003-2202-5279 ABSTRACT Deinococcus bacteria are famous for their extreme resistance to ionising radiation and other DNA damage- and oxidative stress-generating agents. More than a hundred genes have been reported to contribute to resistance to radiation, desiccation and/or oxidative stress in Deinococcus radiodurans. These encode proteins involved in DNA repair, oxidative stress defence, regulation and proteins of yet unknown function or with an extracytoplasmic location. Here, we analysed the conservation of radiation resistance-associated proteins in other radiation-resistant Deinococcus species. Strikingly, homologues of dozens of these proteins are absent in one or more Deinococcus species. -

Supplemental Material-PDF
List of supplementary tables and figures. Table S1 – Strains sequenced and assembly information (n=72) Table S2 – Genetic clusters obtained using BAPS Table S3 – Genes commonly found to be under positive selection in the three subspecies (n=14) Table S4 - Recombining genes in X. fastidiosa subsp. pauca strains 9a5c and U24D (Citrus) Table S5 – Recombination genes for X. fastidiosa 32 (Coffee) Table S6 - Genes found to be under selection in each subspecies Table S7 – Recombining genes for each X. fastidiosa subspecies Table S8 – Genes found in the accessory genome of each subspecies Figure S1 - Phylogeny of X. fastidiosa strains (n=72) and pan-genome visualization Figure S2 - Detection of temporal signal in the dataset Figure S3 - Rate comparison amongst subspecies. The boxplot (A) was constructed my merging the rate of evolution of each branch (internal and external) of the X. fastidiosa subsp. fastidiosa, pauca and multiplex clades together (obtained though rate-dating) as illustrated by the colored frames (B) Figure S4 - Comparison between clonalFrameML and FastGEAR. (A) ClonalFrameML output, (B) FastGEAR output, (C) comparison between the genes found to be recombining between the two algorithms that were mapped to the X. fastidiosa reference genome Table S1 – Strains sequenced and assembly information (n=72). Strain names in parentheses represent synonyms. Name N50 L50 Total length Country Host Subspecies Isolation date Accession CO33 176,135 6 2,681,926 Italy Coffea arabica ? 2015 SAMN04100274 MUL0034 2,642,186 1 2,666,577 USA Morus alba morus 2003 SAMN03081485 Mul-MD 134,146 6 2,520,555 USA Morus alba morus 2011 SAMN02630185 Ann-1 44,355 22 2,729,755 USA Nerium oleander sandyi SAMN03081476 Xf_ATCC_35879 500,648 2 2,522,328 USA Vitis vinifera fastidiosa 1987 SAMN02997312 DSM10026 89,916 11 2,431,652 USA Vitis vinifera fastidiosa 1987 SAMN05660380 EB92-1 44,355 22 2,729,755 USA Sambucus sp. -

Early Stages of Host Invasion by Pseudomonas Aeruginosa and Effect of Cyclic Diguanylate Signaling
EARLY STAGES OF HOST INVASION BY PSEUDOMONAS AERUGINOSA AND EFFECT OF CYCLIC DIGUANYLATE SIGNALING AYSHWARYA RAVICHANDRAN A THESIS SUBMITTED FOR THE DEGREE OF DOCTOR OF PHILOSOPHY DEPARTMENT OF BIOLOGICAL SCIENCES NATIONAL UNIVERSITY OF SINGAPORE 2010 ACKNOWLEDGEMENTS I express my heartfelt gratitude to my supervisor, A/P Sanjay Swarup for his constant guidance and supervision throughout the period of this project. I sincerely thank National University of Singapore for providing me with Research Scholarship to complete this project. I would also like to thank Research Centre for Excellence in Mechanobiology for funding part of this study and support. I am extremely thankful to Dr. Yasushi Ishihama , Keio University, Japan for performing phoshoproteome analysis on our samples without which my publication would not have been possible. Helium-ion imaging was conducted under Dr. Daniel Pickard and I am thankful for his guidance and facility. I extend my sincere gratitude to Dr. Gerard Michel , Centre National de la Recherche Scientifique , France for his kind gesture of sending antibodies and guidance in P. aeruginosa type II secretion system- related experiments. I would also like to thank Dr. Zhang Lian-Hui for providing workspace in his laboratory during the initial stages of this project and Dr. Ganesh Anand for his valuable scientific discussions time-to-time. I express my thanks to Malarmathy Ramachandran and Karen Lam who have been very instrumental in helping me with optimization of experimental methods used in this study. I express my gratitude to Protein and Proteomics centre for their mass spectrometry services, Electron microscopy and the confocal microscopy facilities at the Faculty of Medicine, and the Electron microscopy facility at Department of Biological Sciences. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Polyamines Mitigate Antibiotic Inhibition of A.Actinomycetemcomitans Growth
Polyamines Mitigate Antibiotic Inhibition of A.actinomycetemcomitans Growth THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Allan Wattimena Graduate Program in Dentistry The Ohio State University 2017 Master's Examination Committee: Dr John Walters, Advisor Dr Purnima Kumar Dr Sara Palmer Dr Shareef Dabdoub Copyright by Allan Wattimena 2017 Abstract Polyamines are ubiquitous polycationic molecules that are present in all prokaryotic and eukaryotic cells. They are the breakdown products of amino acids and are important modulators of cell growth, stress and cell proliferation. Polyamines are present in higher concentrations in the periodontal pocket and may affect antibiotic resistance of bacterial biofilms. The effect of polyamines was investigated with amoxicillin (AMX), azithromycin (AZM) and doxycycline (DOX) on the growth of Aggregatibacter actinomycetemcomitans (A.a.) Y4 strain. Bacteria were grown in brain heart infusion broth under the following conditions: 1) A.a. only, 2) A.a. + antibiotic, 3) A.a. + antibiotic + polyamine mix (1.4mM putrescine, 0.4mM spermidine, 0.4mM spermine). Growth curve analysis, MIC determination and metatranscriptomic analysis were carried out. The presence of exogenous polyamines produced a small, but significant increase in growth of A.a. Polyamines mitigated the inhibitory effect of AMX, AZM and DOX on A.a. growth. Metatranscriptomic analysis revealed differing transcriptomic profiles when comparing AMX and AZM in the presence of polyamines. Polyamines produced a transient mitigation of AMX inhibition, but did not have a significant effect on gene transcription. Many gene transcription changes were seen when polyamines were in the presence of AZM. -
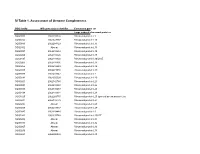
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Characterization of Swarm-Colony Development Reveals the Release of a Distinct Cell Type Facilitating Dissemination of Vibrio Parahaemolyticus
Characterization of swarm-colony development reveals the release of a distinct cell type facilitating dissemination of Vibrio parahaemolyticus Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) dem Fachbereich Biologie der Philipps-Universität Marburg vorgelegt von Carolina Duarte de Freitas aus Lisboa, Portugal Marburg (Lahn), im Juli 2019 Originaldokument gespeichert auf dem Publikationsserver der Philipps-Universität Marburg http://archiv.ub.uni-marburg.de Dieses Werk bzw. Inhalt steht unter einer Creative Commons Namensnennung Keine kommerzielle Nutzung Weitergabe unter gleichen Bedingungen 3.0 Deutschland Lizenz. Die vollständige Lizenz finden Sie unter: http://creativecommons.org/licenses/by-nc-sa/3.0/de/ Die Untersuchungen zur vorliegenden Arbeit wurden von Oktober 2015 bis Juli 2019 am Max-Planck-Institut für Terrestrische Mikrobiologie unter der Leitung von Dr. Simon Ringgaard durchgeführ. Vom Fachbereich Biologie der Philipps-Universität Marburg als Dissertation angenommen am: Erstgutachter: Dr. Simon Ringgaard Zweitgutachter: Prof. Dr. Martin Thanbichler Weitere Mitglieder der Prüfungskommission: Prof. Dr. Knut Drescher Prof. Dr. Michael Bölker Tag der mündlichen Prüfung am: 24.09.2019 Die während der Promotien erzielten Erbebnisse wurden zum Teil in folgenden Originalpublikationen veröffentlicht: Muraleedharan, S., Freitas, C., Mann, P., Glatter, T. and Ringgaard, S. (2018). A cell length-dependent transition in MinD-dynamics promotes a switch in division-site placement and preservation of proliferating elongated Vibrio parahaemolyticus swarmer cells. Molecular Microbiology 109(3): 365-384 Unpublished work: Freitas, C., Glatter, T. and Ringgaard, S. The release of a distinct cell type from swarm colonies facilitates dissemination of Vibrio parahaemolyticus in the environment. Under review, The ISME Journal To my family Table of contents Table of contents Contents Table of contents.............................................................................................................