Essential Psychopharmacology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

From Inverse Agonism to 'Paradoxical Pharmacology' Richard A
International Congress Series 1249 (2003) 27-37 From inverse agonism to 'Paradoxical Pharmacology' Richard A. Bond*, Kenda L.J. Evans, Zsirzsanna Callaerts-Vegh Department of Pharmacological and Pharmaceutical Sciences, University of Houston, 521 Science and Research Bldg 2, 4800 Caltioun, Houston, TX 77204-5037, USA Received 16 April 2003; accepted 16 April 2003 Abstract The constitutive or spontaneous activity of G protein-coupled receptors (GPCRs) and compounds acting as inverse agonists is a recent but well-established phenomenon. Dozens of receptor subtypes for numerous neurotransmitters and hormones have been shown to posses this property. However, do to the apparently low percentage of receptors in the spontaneously active state, the physiologic relevance of these findings remains questionable. The possibility that the reciprocal nature of the effects of agonists and inverse agonists may extend to cellular signaling is discussed, and that this may account for the beneficial effects of certain p-adrenoceptor inverse agonists in the treatment of heart failure. © 2003 Elsevier Science B.V. All rights reserved. Keywords. Inverse agonism; GPCR; Paradoxical pharmacology 1. Brief history of inverse agonism at G protein-coupled receptors For approximately three-quarters of a century, ligands that interacted with G protein- coupled receptors (GPCRs) were classified either as agonists or antagonists. Receptors were thought to exist in a single quiescent state that could only induce cellular signaling upon agonist binding to the receptor to produce an activated state of the receptor. In this model, antagonists had no cellular signaling ability on their own, but did bind to the receptor and prevented agonists from being able to bind and activate the receptor. -

Making Sense of Pharmacology: Inverse Agonism and Functional Selectivity Kelly A
International Journal of Neuropsychopharmacology (2018) 21(10): 962–977 doi:10.1093/ijnp/pyy071 Advance Access Publication: August 6, 2018 Review review Making Sense of Pharmacology: Inverse Agonism and Functional Selectivity Kelly A. Berg and William P. Clarke Department of Pharmacology, University of Texas Health, San Antonio, Texas. Correspondence: William P. Clarke, PhD, Department of Pharmacology, Mail Stop 7764, UT Health at San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229 ([email protected]). Abstract Constitutive receptor activity/inverse agonism and functional selectivity/biased agonism are 2 concepts in contemporary pharmacology that have major implications for the use of drugs in medicine and research as well as for the processes of new drug development. Traditional receptor theory postulated that receptors in a population are quiescent unless activated by a ligand. Within this framework ligands could act as agonists with various degrees of intrinsic efficacy, or as antagonists with zero intrinsic efficacy. We now know that receptors can be active without an activating ligand and thus display “constitutive” activity. As a result, a new class of ligand was discovered that can reduce the constitutive activity of a receptor. These ligands produce the opposite effect of an agonist and are called inverse agonists. The second topic discussed is functional selectivity, also commonly referred to as biased agonism. Traditional receptor theory also posited that intrinsic efficacy is a single drug property independent of the system in which the drug acts. However, we now know that a drug, acting at a single receptor subtype, can have multiple intrinsic efficacies that differ depending on which of the multiple responses coupled to a receptor is measured. -

Cannabinoid-1 Receptor Inverse Agonists: Current Understanding of Mechanism of Action and Unanswered Questions
International Journal of Obesity (2009) 33, 947–955 & 2009 Macmillan Publishers Limited All rights reserved 0307-0565/09 $32.00 www.nature.com/ijo REVIEW Cannabinoid-1 receptor inverse agonists: current understanding of mechanism of action and unanswered questions TM Fong1 and SB Heymsfield2 1Merck Research Laboratories, Department of Metabolic Disorders, Rahway, NJ, USA and 2Merck Research Laboratories, Global Center of Scientific Affairs, Rahway, NJ, USA Rimonabant and taranabant are two extensively studied cannabinoid-1 receptor (CB1R) inverse agonists. Their effects on in vivo peripheral tissue metabolism are generally well replicated. The central nervous system site of action of taranabant or rimonabant is firmly established based on brain receptor occupancy studies. At the whole-body level, the mechanism of action of CB1R inverse agonists includes a reduction in food intake and an increase in energy expenditure. At the tissue level, fat mass reduction, liver lipid reduction and improved insulin sensitivity have been shown. These effects on tissue metabolism are readily explained by CB1R inverse agonist acting on brain CB1R and indirectly influencing the tissue metabolism through the autonomic nervous system. It has also been hypothesized that rimonabant acts directly on adipocytes, hepatocytes, pancreatic islets or skeletal muscle in addition to acting on brain CB1R, although strong support for the contribution of peripherally located CB1R to in vivo efficacy is still lacking. This review will carefully examine the published literature -

Different Inverse Agonist Activities of P»-Adrenergic Receptor Antagonists—Pharmacological Characterization and Therapeutical
International Congress Series 1249 (2003) 39-53 Different inverse agonist activities of p»-adrenergic receptor antagonists—pharmacological characterization and therapeutical implications in the treatment of chronic heart failure Christoph Maack*, Michael Bòhm Medizinische Klinik und Poliklinik fiir Innere Medizin III, Universitat des Saarlandes, 66421 Homburg/Saai; Germany Received 16 April 2003; accepted 16 April 2003 Abstract The treatment of chronic heart failure with most p-adrenergic receptor (p-AR) antagonists leads to an improvement of symptoms and left ventricular function. However, only metoprolol, bisoprolol and carvedilol have been shown to reduce mortality in these patients. Bucindolol did not reduce mortality and xamoterol even increased it. These differences may be related to different inverse agonist or partial agonist activity of p-AR antagonists. This review focusses on the determination of different intrinsic activity of the mentioned p-AR antagonists in the human myocardium. Furthermore, the clinical impact of these differences is examined. In this regard, the effect of the different p-AR antagonists on p-AR regulation, minimum heart rate and exercise tolerance, as well as prognosis, is highlighted. It is concluded that the degree of inverse agonism of a p-AR antagonist determines the degree of p-AR resensitization, reduction of minimum heart rate, improvement of exercise tolerance and possibly also improvement of prognosis of patients with chronic heart failure. © 2003 Elsevier Science B.V. All rights reserved. Key\vords: Inverse agonism; p-adrenergic receptors; p-blockers, Heart faitee * Corresponding author. Current address: The Johns Hopkins University, Institute of Molecular Cardio- biology, Division of Cardiology, 720 Rutland Ave., 844 Ross Bldg., Baltimore, MD 21205-2195, USA. -

Pharmacokinetics and Pharmacology of Drugs Used in Children
Drug and Fluid Th erapy SECTION II Pharmacokinetics and Pharmacology of Drugs Used CHAPTER 6 in Children Charles J. Coté, Jerrold Lerman, Robert M. Ward, Ralph A. Lugo, and Nishan Goudsouzian Drug Distribution Propofol Protein Binding Ketamine Body Composition Etomidate Metabolism and Excretion Muscle Relaxants Hepatic Blood Flow Succinylcholine Renal Excretion Intermediate-Acting Nondepolarizing Relaxants Pharmacokinetic Principles and Calculations Atracurium First-Order Kinetics Cisatracurium Half-Life Vecuronium First-Order Single-Compartment Kinetics Rocuronium First-Order Multiple-Compartment Kinetics Clinical Implications When Using Short- and Zero-Order Kinetics Intermediate-Acting Relaxants Apparent Volume of Distribution Long-Acting Nondepolarizing Relaxants Repetitive Dosing and Drug Accumulation Pancuronium Steady State Antagonism of Muscle Relaxants Loading Dose General Principles Central Nervous System Effects Suggamadex The Drug Approval Process, the Package Insert, and Relaxants in Special Situations Drug Labeling Opioids Inhalation Anesthetic Agents Morphine Physicochemical Properties Meperidine Pharmacokinetics of Inhaled Anesthetics Hydromorphone Pharmacodynamics of Inhaled Anesthetics Oxycodone Clinical Effects Methadone Nitrous Oxide Fentanyl Environmental Impact Alfentanil Oxygen Sufentanil Intravenous Anesthetic Agents Remifentanil Barbiturates Butorphanol and Nalbuphine 89 A Practice of Anesthesia for Infants and Children Codeine Antiemetics Tramadol Metoclopramide Nonsteroidal Anti-infl ammatory Agents 5-Hydroxytryptamine -

Equilibrium Assays Are Required to Accurately Characterize the Activity Profiles of Drugs
Molecular Pharmacology Fast Forward. Published on June 28, 2018 as DOI: 10.1124/mol.118.112573 This article has not been copyedited and formatted. The final version may differ from this version. MOL112573 Equilibrium Assays are Required to Accurately Characterize the Activity Profiles of Drugs Modulating Gq-Coupled GPCRs Sara Bdioui, Julien Verdi, Nicolas Pierre, Eric Trinque, Thomas Roux, Terry Kenakin SB, JV, NP, ET, TR: Cisbio Bioassays, 30200 Codolet, France TK : Department of Pharmacology, University of North Carolina School of Medicine, Chapel Hill, North Carolina Downloaded from molpharm.aspetjournals.org at ASPET Journals on September 26, 2021 1 Molecular Pharmacology Fast Forward. Published on June 28, 2018 as DOI: 10.1124/mol.118.112573 This article has not been copyedited and formatted. The final version may differ from this version. MOL112573 RUNNING (SHORT) TITLE Functional assays for Gq Protein activating Receptor Ligands Corresponding author: Terry Kenakin Ph.D. Professor, Department of Pharmacology University of North Carolina School of Medicine Downloaded from 120 Mason Farm Road Room 4042 Genetic Medicine Building, CB# 7365 molpharm.aspetjournals.org Chapel Hill, NC 27599-7365 Phone: 919-962-7863 Fax: 919-966-7242 or 5640 Email: [email protected] at ASPET Journals on September 26, 2021 Number of text pages: 56 Number of tables: 0 Number of figures: 13 Number of references: 33 Number of words in the Abstract: 233 Number of words in the Introduction: 599 Number of words in the Discussion: 1497 2 Molecular Pharmacology Fast Forward. Published on June 28, 2018 as DOI: 10.1124/mol.118.112573 This article has not been copyedited and formatted. -

Immunomodulatory Potential of Cannabidiol in Multiple Sclerosis: a Systematic Review
Journal of Neuroimmune Pharmacology https://doi.org/10.1007/s11481-021-09982-7 INVITED REVIEW Immunomodulatory Potential of Cannabidiol in Multiple Sclerosis: a Systematic Review Alessia Furgiuele1 & Marco Cosentino1 & Marco Ferrari1 & Franca Marino1 Received: 16 December 2020 /Accepted: 6 January 2021 # The Author(s) 2021 Abstract Multiple sclerosis (MS) is the most common chronic autoimmune disease of the central nervous system. Efficacy of treatments for MS is associated with risk of adverse effects, and effective and well-tolerated drugs remain a major unmet need. Cannabis (Cannabis sativa L., fam. Cannabaceae) and cannabinoids are popular among MS patients to treat spasticity and pain. Cannabinoids are endowed with remarkable immunomodulating properties, and in particular the non-psychotropic cannabinoid cannabidiol (CBD) is increasingly recognized as anti-inflammatory and immunosuppressive, nevertheless with excellent toler- ability even at high doses. In this systematic review, we retrieved and critically evaluated available evidence regarding the immune and disease-modifying effects of CBD in experimental autoimmune encephalomyelitis (EAE) and in MS. Evidence in rodent models of EAE strongly supports CBD as effective, while clinical evidence is still limited and usually negative, due to paucity of studies and possibly to the use of suboptimal dosing regimens. Better characterization of targets acted upon by CBD in MS should be obtained in ex vivo/in vitro studies in human immune cells, and higher doses should be tested in well-designed clinical trials with clinically relevant efficacy endpoints. Keywords Multiple sclerosis . Experimental autoimmune encephalomyelitis . Cannabidiol . Immunomodulation Introduction (PRMS), occurring in fewer than 5% of patients (Dobson and Giovannoni 2019; Reich et al. -

International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology
0031-6997/03/5504-597–606$7.00 PHARMACOLOGICAL REVIEWS Vol. 55, No. 4 Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 30404/1114803 Pharmacol Rev 55:597–606, 2003 Printed in U.S.A International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology RICHARD R. NEUBIG, MICHAEL SPEDDING, TERRY KENAKIN, AND ARTHUR CHRISTOPOULOS Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (R.R.N.); Institute de Recherches Internationales Servier, Neuilly sur Seine, France (M.S.); Systems Research, GlaxoSmithKline Research and Development, Research Triangle Park, North Carolina (T.K.); and Department of Pharmacology, University of Melbourne, Parkville, Australia (A.C.) Abstract ............................................................................... 597 I. Introduction............................................................................ 597 II. Working definition of a receptor .......................................................... 598 III. Use of drugs in definition of receptors or of signaling pathways ............................. 598 A. The expression of amount of drug: concentration and dose ............................... 598 Downloaded from 1. Concentration..................................................................... 598 2. Dose. ............................................................................ 598 B. General terms used to describe drug action ........................................... -

Effects of the Kappa-Opioid Receptor Agonist, U69593, on the Development of Sensitization and on the Maintenance of Cocaine Self
Effects of the Kappa-opioid Receptor Agonist, U69593, on the Development of Sensitization and on the Maintenance of Cocaine Self-administration Susan Schenk, Ph.D., Brian Partridge, M.S., and Toni S. Shippenberg, Ph.D. Previous studies showed that prior administration of with each self-administered cocaine infusion for one group kappa-opioid agonists decreased the development of whereas responding of another group was reinforced by a sensitization to some of the behavioral effects of cocaine. The cocaine infusion alone. On the test day, pretreatment with present study sought to determine whether the development U69593 (0.32 mg/kg) decreased responding during each of sensitization to cocaine’s reinforcing effects was also hour of the 10 hr session for the group that was reinforced sensitive to antagonism by kappa-opioid agonists. During a with cocaine plus the cocaine-associated stimulus. U69593 pretreatment phase, the kappa-opioid agonist, U69593 (0.0 failed to produce a long-lasting disruption of cocaine self- or 0.32 mg/kg) was administered prior to (1) 2 daily administration for rats that were trained and tested without injections of cocaine (0.0 or 20.0 mg/kg), or (2) cocaine or the cocaine-associated stimulus. These data suggest that the saline administered via a yoking procedure. Cocaine acquisition and maintenance of cocaine self-administration pretreatment decreased the latency to acquisition of cocaine are differentially sensitive to manipulations of kappa-opioid self-administration. However, prior administration of systems. Further, the disruption of cocaine self- U69593 during the pretreatment phase failed to attenuate administration by U69593 may be due to interactions with the development of this sensitized response to cocaine’s mechanisms that underlie facilitative effects of stimuli that reinforcing effect. -
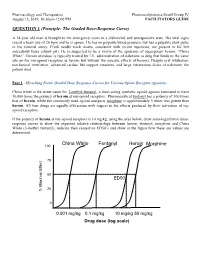
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined. -

Introduction to Pharmacodynamics Reza Karimi 6
CHAPTER Introduction to Pharmacodynamics Reza Karimi 6 1. Understand the physiology behind the gastrointestinal tract and the route of oral drug administration and VES physiological influences on pharmacodynamics. I 2. Understand the dynamics and functions of the major signal transduction systems and their different biomedi- cal and biological responses in regard to receptor–ligand interactions. 3. Learn about the dynamics and mathematical expressions behind receptor–ligand interactions. OBJECT 4. Understand dose–response relationships and factors that affect a pharmacological response. 5. Learn about agonistic, antagonistic, and partial agonistic binding of drugs to receptors. 6. Learn about different concepts such as addition, synergism, and potentiation that lead to an enhancement effect of drugs. 7. List a few regulatory mechanisms for receptors. 8. Implement a series of Learning Bridge assignments at your experiential sites to bridge your didactic learning with your experiential experiences. 1. cAMP: cyclic adenosine 3' ,5''-monophosphate; a second messenger that plays an important role in signal NS transduction. IO T 2. cGMP: cyclic guanosine 3' ,5''monophosphate; a second messenger that plays an important role in signal I N transduction. I 3. Dose–response relationship: when an endogenous or exogenous ligand binds to a receptor and produces a EF D pharmacological effect. The effect can approach a maximum value (also called Emax) in which a further increase in the ligand concentration does not produce any higher response. 4. Efficacy: the ability of a drug to produce a pharmacological response when it interacts with its receptor. 5. First-pass metabolism: a type of metabolism in which drugs that are absorbed by the gastrointestinal tract go through the portal vein to the liver and are metabolized there before they are distributed to the general ERMS AND AND ERMS circulation. -

The Pharmacology of an Agonist Medication to Treat Stimulant Use Disorder
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2017 The Pharmacology of an Agonist Medication to Treat Stimulant Use Disorder Amy Johnson johnsonar24 Follow this and additional works at: https://scholarscompass.vcu.edu/etd © Amy Johnson Downloaded from https://scholarscompass.vcu.edu/etd/5177 This Dissertation is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. © Amy R. Johnson 2017 All Rights Reserved The Pharmacology of an Agonist Medication to Treat Stimulant Use Disorder A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy at Virginia Commonwealth University by Amy R. Johnson Bachelor of Science, University of Wisconsin - Eau Claire, 2013 Advisor: S. Stevens Negus, PhD Professor of Pharmacology and Toxicology Virginia Commonwealth University Virginia Commonwealth University Richmond, VA November, 2017 Acknowledgements Thank you to all the people who made this dissertation possible. I would like to thank my family for their support and belief in me. Many thanks to my dissertation advisor, Steve Negus, for his guidance, knowledge, patience, and encouragement. Thank you to Katherine Nicholson and Matthew Banks, each of whom guided me through a different procedure through the course of my research and served on my committee. Thanks to my remaining committee members, Lori Keyser-Marcus and Jose Eltit, for their time and commitment to my project. Thank you to my entire committee for helping to develop and mold me as a scientist.