Inside-Out Neuropharmacology of Nicotinic Drugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacokinetics and Pharmacology of Drugs Used in Children
Drug and Fluid Th erapy SECTION II Pharmacokinetics and Pharmacology of Drugs Used CHAPTER 6 in Children Charles J. Coté, Jerrold Lerman, Robert M. Ward, Ralph A. Lugo, and Nishan Goudsouzian Drug Distribution Propofol Protein Binding Ketamine Body Composition Etomidate Metabolism and Excretion Muscle Relaxants Hepatic Blood Flow Succinylcholine Renal Excretion Intermediate-Acting Nondepolarizing Relaxants Pharmacokinetic Principles and Calculations Atracurium First-Order Kinetics Cisatracurium Half-Life Vecuronium First-Order Single-Compartment Kinetics Rocuronium First-Order Multiple-Compartment Kinetics Clinical Implications When Using Short- and Zero-Order Kinetics Intermediate-Acting Relaxants Apparent Volume of Distribution Long-Acting Nondepolarizing Relaxants Repetitive Dosing and Drug Accumulation Pancuronium Steady State Antagonism of Muscle Relaxants Loading Dose General Principles Central Nervous System Effects Suggamadex The Drug Approval Process, the Package Insert, and Relaxants in Special Situations Drug Labeling Opioids Inhalation Anesthetic Agents Morphine Physicochemical Properties Meperidine Pharmacokinetics of Inhaled Anesthetics Hydromorphone Pharmacodynamics of Inhaled Anesthetics Oxycodone Clinical Effects Methadone Nitrous Oxide Fentanyl Environmental Impact Alfentanil Oxygen Sufentanil Intravenous Anesthetic Agents Remifentanil Barbiturates Butorphanol and Nalbuphine 89 A Practice of Anesthesia for Infants and Children Codeine Antiemetics Tramadol Metoclopramide Nonsteroidal Anti-infl ammatory Agents 5-Hydroxytryptamine -

International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology
0031-6997/03/5504-597–606$7.00 PHARMACOLOGICAL REVIEWS Vol. 55, No. 4 Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 30404/1114803 Pharmacol Rev 55:597–606, 2003 Printed in U.S.A International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology RICHARD R. NEUBIG, MICHAEL SPEDDING, TERRY KENAKIN, AND ARTHUR CHRISTOPOULOS Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (R.R.N.); Institute de Recherches Internationales Servier, Neuilly sur Seine, France (M.S.); Systems Research, GlaxoSmithKline Research and Development, Research Triangle Park, North Carolina (T.K.); and Department of Pharmacology, University of Melbourne, Parkville, Australia (A.C.) Abstract ............................................................................... 597 I. Introduction............................................................................ 597 II. Working definition of a receptor .......................................................... 598 III. Use of drugs in definition of receptors or of signaling pathways ............................. 598 A. The expression of amount of drug: concentration and dose ............................... 598 Downloaded from 1. Concentration..................................................................... 598 2. Dose. ............................................................................ 598 B. General terms used to describe drug action ........................................... -

Effects of the Kappa-Opioid Receptor Agonist, U69593, on the Development of Sensitization and on the Maintenance of Cocaine Self
Effects of the Kappa-opioid Receptor Agonist, U69593, on the Development of Sensitization and on the Maintenance of Cocaine Self-administration Susan Schenk, Ph.D., Brian Partridge, M.S., and Toni S. Shippenberg, Ph.D. Previous studies showed that prior administration of with each self-administered cocaine infusion for one group kappa-opioid agonists decreased the development of whereas responding of another group was reinforced by a sensitization to some of the behavioral effects of cocaine. The cocaine infusion alone. On the test day, pretreatment with present study sought to determine whether the development U69593 (0.32 mg/kg) decreased responding during each of sensitization to cocaine’s reinforcing effects was also hour of the 10 hr session for the group that was reinforced sensitive to antagonism by kappa-opioid agonists. During a with cocaine plus the cocaine-associated stimulus. U69593 pretreatment phase, the kappa-opioid agonist, U69593 (0.0 failed to produce a long-lasting disruption of cocaine self- or 0.32 mg/kg) was administered prior to (1) 2 daily administration for rats that were trained and tested without injections of cocaine (0.0 or 20.0 mg/kg), or (2) cocaine or the cocaine-associated stimulus. These data suggest that the saline administered via a yoking procedure. Cocaine acquisition and maintenance of cocaine self-administration pretreatment decreased the latency to acquisition of cocaine are differentially sensitive to manipulations of kappa-opioid self-administration. However, prior administration of systems. Further, the disruption of cocaine self- U69593 during the pretreatment phase failed to attenuate administration by U69593 may be due to interactions with the development of this sensitized response to cocaine’s mechanisms that underlie facilitative effects of stimuli that reinforcing effect. -
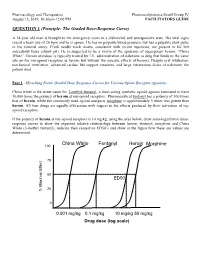
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined. -

Introduction to Pharmacodynamics Reza Karimi 6
CHAPTER Introduction to Pharmacodynamics Reza Karimi 6 1. Understand the physiology behind the gastrointestinal tract and the route of oral drug administration and VES physiological influences on pharmacodynamics. I 2. Understand the dynamics and functions of the major signal transduction systems and their different biomedi- cal and biological responses in regard to receptor–ligand interactions. 3. Learn about the dynamics and mathematical expressions behind receptor–ligand interactions. OBJECT 4. Understand dose–response relationships and factors that affect a pharmacological response. 5. Learn about agonistic, antagonistic, and partial agonistic binding of drugs to receptors. 6. Learn about different concepts such as addition, synergism, and potentiation that lead to an enhancement effect of drugs. 7. List a few regulatory mechanisms for receptors. 8. Implement a series of Learning Bridge assignments at your experiential sites to bridge your didactic learning with your experiential experiences. 1. cAMP: cyclic adenosine 3' ,5''-monophosphate; a second messenger that plays an important role in signal NS transduction. IO T 2. cGMP: cyclic guanosine 3' ,5''monophosphate; a second messenger that plays an important role in signal I N transduction. I 3. Dose–response relationship: when an endogenous or exogenous ligand binds to a receptor and produces a EF D pharmacological effect. The effect can approach a maximum value (also called Emax) in which a further increase in the ligand concentration does not produce any higher response. 4. Efficacy: the ability of a drug to produce a pharmacological response when it interacts with its receptor. 5. First-pass metabolism: a type of metabolism in which drugs that are absorbed by the gastrointestinal tract go through the portal vein to the liver and are metabolized there before they are distributed to the general ERMS AND AND ERMS circulation. -

The Pharmacology of an Agonist Medication to Treat Stimulant Use Disorder
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2017 The Pharmacology of an Agonist Medication to Treat Stimulant Use Disorder Amy Johnson johnsonar24 Follow this and additional works at: https://scholarscompass.vcu.edu/etd © Amy Johnson Downloaded from https://scholarscompass.vcu.edu/etd/5177 This Dissertation is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. © Amy R. Johnson 2017 All Rights Reserved The Pharmacology of an Agonist Medication to Treat Stimulant Use Disorder A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy at Virginia Commonwealth University by Amy R. Johnson Bachelor of Science, University of Wisconsin - Eau Claire, 2013 Advisor: S. Stevens Negus, PhD Professor of Pharmacology and Toxicology Virginia Commonwealth University Virginia Commonwealth University Richmond, VA November, 2017 Acknowledgements Thank you to all the people who made this dissertation possible. I would like to thank my family for their support and belief in me. Many thanks to my dissertation advisor, Steve Negus, for his guidance, knowledge, patience, and encouragement. Thank you to Katherine Nicholson and Matthew Banks, each of whom guided me through a different procedure through the course of my research and served on my committee. Thanks to my remaining committee members, Lori Keyser-Marcus and Jose Eltit, for their time and commitment to my project. Thank you to my entire committee for helping to develop and mold me as a scientist. -

Are All GLP-1 Agonists Equal in the Treatment of Type 2 Diabetes?
6 181 M A Nauck and J J Meier GLP-1 receptor agonist 181:6 R211–R234 Review comparison MANAGEMENT OF ENDOCRINE DISEASE Are all GLP-1 agonists equal in the treatment of type 2 diabetes? Correspondence should be addressed Michael A Nauck and Juris J Meier to M A Nauck Diabetes Division, St. Josef-Hospital, Ruhr-University of Bochum, Bochum, Germany Email [email protected] Abstract GLP-1, a peptide hormone secreted from the gut, stimulating insulin and suppressing glucagon secretion was identified as a parent compound for novel treatments of diabetes, but was degraded (dipeptidyl peptidase-4) and eliminated (mainly by kidneys) too fast (half-life 1–2 min) to be useful as a therapeutic agent. GLP-1 receptor agonist has been used to treat patients with type 2 diabetes since 2007, when exenatide (twice daily) was approved in 2007. Compounds with longer duration of action (once daily, once weekly) and with increasingly better efficacy with respect to glycaemic control and body weight reduction have been developed, and in a recent ADA/EASD consensus statement, were recommended as the first injectable diabetes therapy after failure of oral glucose-lowering medications. Most GLP-1 receptor agonists (lixisenatide q.d., liraglutide q.d., exenatide q.w., dulaglutide q.w., albiglutide q.w., semaglutide q.w., all for s.c. injection, and the first oral preparation, oral semaglutide) have been examined in cardiovascular outcomes studies. Beyond proving their safety in vulnerable patients, most of whom had pre-existing heart disease, liraglutide, semaglutide, albiglutide, and dulaglutide reduced the time to first major adverse cardiovascular events (non-fatal myocardial infarction and stroke, cardiovascular death). -

Update on the Role of Alpha-Agonists in Glaucoma Management
Experimental Eye Research 93 (2011) 271e283 Contents lists available at ScienceDirect Experimental Eye Research journal homepage: www.elsevier.com/locate/yexer Update on the role of alpha-agonists in glaucoma management Stella Arthur, Louis B. Cantor* Eugene and Marilyn Glick Eye Institute, Department of Ophthalmology, Indiana University School of Medicine, 702 Rotary Circle, Indianapolis, IN 46202, USA article info abstract Article history: Glaucoma is the second most common cause of world blindness (following cataract) with estimated Received 30 November 2010 cases reaching 79.6 million by 2020. Although the etiology of glaucoma is multi-factorial, intraocular Accepted in revised form 4 April 2011 pressure (IOP) is the only modifiable factor in glaucoma management proven to alter the natural course Available online 20 April 2011 of the disease. Among various classes of IOP-lowering medications currently available, alpha-adrenergic receptor agonists are used either as monotherapy, as second-line therapy, or in fixed combination with Keywords: beta-blockers. Non-selective adrenergic agonists such as epinephrine and dipivefrin are infrequently glaucoma used today for the treatment of glaucoma or ocular hypertension, and have been replaced by the alpha-2- alpha-agonists monotherapy selective agonists. The use of apraclonidine for IOP reduction in glaucoma or OHT is limited due to a high fixed combinations rate of follicular conjunctivitis. The alpha-2-selective agonist in use today is brimonidine. The brimo- neuroprotection nidineepurite formulations are preferred to brimonidineebenzalkonium chloride (BAC) formulations ocular blood flow due better tolerability while maintaining similar efficacy. Brimonidine is also effective when used in safety and efficacy combination with a beta-blocker. -

Essential Psychopharmacology
CONTENTS Preface vii Chapter 1 Principles of Chemical Neurotransmission 1 Chapter 2 Receptors and Enzymes as the Targets of Drug Action 35 Chapter 3 Special Properties of Receptors 77 Chapter 4 Chemical Neurotransmission as the Mediator of Disease Actions 99 Chapter 5 Depression and Bipolar Disorders 135 Chapter 6 Classical Antidepressants, Serotonin Selective Reuptake Inhibitors, and. Noradrenergic Reuptake Inhibitors 199 Chapter 7 Newer Antidepressants and Mood Stabilizers 245 Chapter 8 Anxiolytics and Sedative-Hypnotics 297 Chapter 9 Drug Treatments for Obsessive-Compulsive Disorder, Panic Disorder, and Phobic Disorders 335 xi xii Contens Chapter 10 Psychosis and Schizophrenia 365 Chapter 11 Antipsychotic Agents 401 Chapter 12 Cognitive Enhancers 459 Chapter 13 Psychopharmacology of Reward and Drugs of Abuse 499 Chapter 14 Sex-Specific and Sexual Function-Related Psychopharmacology 539 Suggested Reading 569 Index 575 CME Post Tests and Evaluations CHAPTER 3 SPECIAL PROPERTIES OF RECEPTORS I. Multiple receptor subtypes A. Definition and description B. Pharmacological subtyping C. Receptor superfamilies II. Agonists and antagonists A. Antagonists B. Inverse agonists C. Partial agonists D. Light and dark as an analogy for partial agonists III. Allosteric modulation A. Positive allosteric interactions B. Negative allosteric interactions IV. Co-transmission versus allosteric modulation V. Summary The study of receptor psychopharmacology involves understanding not only that receptors are the targets for most of the known drugs but also that they have some very special properties. This chapter will build on the discussion of the general properties of receptors introduced in Chapter 2 and will introduce the reader to some of the special properties of receptors that help explain how they participate in key drug interactions. -

Glucagon GLP-1 Receptor Ligand Binding Assay Figure 9 – Compound Library % Inhibition Distribution
Rapid Screening of a Cell-based Assay for GLP-1 Receptor Using a Natural Product Library Brad Larson1, Nicolas Pierre2, Suzanne Graham2, Jean-Luc Tardieu2, Francois Degorce2, and Peter Banks1 1BioTek Instruments, Inc., Winooski, Vermont, USA • 2Cisbio US, Inc., Bedford, Massachusetts, USA Introduction Methods Natural Product Library Screen Glucagon-like peptide-1 receptor (GLP-1R) is a G-protein coupled receptor that Cell Preparation: Frozen cells are thawed at 37 oC and transferred to a vial containing A total of 384 compounds from plates 1-4 of the Screen-Well Natural Product Library is present in insulin-secreting beta cells. Intact GLP-1(1-37) is produced by post- 5 mL of 1X TLB. The vial is then centrifuged for 5 minutes at 1200xG at 4 oC. were screened in duplicate. Compounds were diluted 1:1000 from the original 100% translational processing of proglucagon precursor and converted into active forms Supernatant is then aspirated and the pellet is resuspended in 2.7 mL of 1X TLB. DMSO stocks as previously described. A no compound (0% inhibition) control was of GLP-1 [(7-36) amide and (7-37)] with N-terminal truncation. Then, active forms of GLP-1 Receptor Red Agonist Preparation: A. K Determination – The 400 nM included on each assay plate. The unlabeled form of the known GLP-1 receptor 1,2 d GLP-1 are deactivated by the further fragmentation with peptidases . concentration of the red agonist is prepared by diluting the stock concentration agonist, Exendin-4, was also included in the library, as well as two known antagonists, Its defining action is augmentation of glucose-induced insulin secretion following in 1X TLB (See kit insert for stock concentration). -

Prediction of Median Lethal Dose by QSAR Method with Their Applications
International Research Journal of Engineering and Technology (IRJET) e-ISSN: 2395 -0056 Volume: 02 Issue: 03 | June-2015 www.irjet.net p-ISSN: 2395-0072 Prediction of Median Lethal Dose by QSAR method with their Applications *Rajendra Kumar Sharma1, Arun Sikarwar2, Rajeeev Sharma3, Pratibha Sharma4 *1 Department of Applied Chemistry, SGSITS, Indore- 452003, M.P., India. 2 Department of Chemistry, Government Home Science College, Hoshangabad- 461001 (M.P.) India. 3 Govt. K. P. College (Vigyan Bhawan), Dewas (M.P.) India. 4 Department of Chemistry, ISLE, IPS Academy, Indore-452012, M.P., India. ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ *Corresponding author’s email address: [email protected] Mob. No. 91-9039541925 ---------------------------------------------------------------------------------------------------------------------------------------- Abstracts: In the present study 49 molecules belonging to biological types of test animal, route of exposure, little on sex cephalosporin antibiotic drugs were selected. Their 2D and of animal and some times slightly on age of test animal. 3D structures were prepared and subjected to Dragon Ratio of LD50 to ED50 is known as Therapatic Index (T.I.) or software for the calculation of various indices viz. topological Therapatic Ratio. (e.g. wiener, randic, schultz, balban, detour etc.), geometrical (TOXICITY)DRUG α LD50 [e.g. (T(N-N), T(N-O), T(N-S)], Quantum mechanical (e.g. 3D → (1) MoRSE, Kier Hall etc.), electronic (polarizability), simple total R R atom no. count indices [(NC) , (NS) ] and combinations of Therapatic Index = LD50 / ED50 various indices like addition, substraction also applied for the → (2) wox wor wox wor study like {(Ss) - (Ss) }, {(Ss) + (Ss) }, For safe use of drug T.I. should be high as far as possible. -

Pharmacology Part 1: Introduction to Pharmaoclogy and Pharmacodynamics
J of Nuclear Medicine Technology, first published online March 29, 2018 as doi:10.2967/jnmt.117.199588 PHARMACOLOGY PART 1: INTRODUCTION TO PHARMACOLOGY AND PHARMACODYNAMICS. Geoffrey M Currie Faculty of Science, Charles Sturt University, Wagga Wagga, Australia. Regis University, Boston, USA. Correspondence: Geoff Currie Faculty of Science Locked Bag 588 Charles Sturt University Wagga Wagga 2678 Australia Telephone: 02 69332822 Facsimile: 02 69332588 Email: [email protected] Foot line: Introduction to Pharmacodynamics 1 Abstract There is an emerging need for greater understanding of pharmacology principles amongst technical staff. Indeed, the responsibility of dose preparation and administration, under any level of supervision, demands foundation understanding of pharmacology. This is true for radiopharmaceuticals, contrast media and pharmaceutical interventions / adjunctive medications. Regulation around the same might suggest a need to embed pharmacology theory in undergraduate education programs and there is a need to disseminate that same foundation understanding to practicing clinicians. Moreover, pharmacology foundations can provide key understanding of the principles that underpin quantitative techniques (e.g. pharmacokinetics). This article is the first in a series of articles that aims to enhance the understanding of pharmacological principles relevant to nuclear medicine. This article will deal with the introductory concepts, terminology and principles that underpin the concepts to be discussed in the remainder of the series. The second article will build on the pharmacodynamic principles examined in this article with a treatment of pharmacokinetics. Article 3 will outline pharmacology relevant to pharmaceutical interventions and adjunctive medications employed in general nuclear medicine, the fourth pharmacology relevant to pharmaceutical interventions and adjunctive medications employed in nuclear cardiology, and the fifth the pharmacology related to contrast media associated with computed tomography (CT) and magnetic resonance imaging (MRI).