Shared Expression of Crassulacean Acid Metabolism (CAM) Genes Predates the Origin of CAM in the Genus Yucca
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Natural Heritage Program List of Rare Plant Species of North Carolina 2016
Natural Heritage Program List of Rare Plant Species of North Carolina 2016 Revised February 24, 2017 Compiled by Laura Gadd Robinson, Botanist John T. Finnegan, Information Systems Manager North Carolina Natural Heritage Program N.C. Department of Natural and Cultural Resources Raleigh, NC 27699-1651 www.ncnhp.org C ur Alleghany rit Ashe Northampton Gates C uc Surry am k Stokes P d Rockingham Caswell Person Vance Warren a e P s n Hertford e qu Chowan r Granville q ot ui a Mountains Watauga Halifax m nk an Wilkes Yadkin s Mitchell Avery Forsyth Orange Guilford Franklin Bertie Alamance Durham Nash Yancey Alexander Madison Caldwell Davie Edgecombe Washington Tyrrell Iredell Martin Dare Burke Davidson Wake McDowell Randolph Chatham Wilson Buncombe Catawba Rowan Beaufort Haywood Pitt Swain Hyde Lee Lincoln Greene Rutherford Johnston Graham Henderson Jackson Cabarrus Montgomery Harnett Cleveland Wayne Polk Gaston Stanly Cherokee Macon Transylvania Lenoir Mecklenburg Moore Clay Pamlico Hoke Union d Cumberland Jones Anson on Sampson hm Duplin ic Craven Piedmont R nd tla Onslow Carteret co S Robeson Bladen Pender Sandhills Columbus New Hanover Tidewater Coastal Plain Brunswick THE COUNTIES AND PHYSIOGRAPHIC PROVINCES OF NORTH CAROLINA Natural Heritage Program List of Rare Plant Species of North Carolina 2016 Compiled by Laura Gadd Robinson, Botanist John T. Finnegan, Information Systems Manager North Carolina Natural Heritage Program N.C. Department of Natural and Cultural Resources Raleigh, NC 27699-1651 www.ncnhp.org This list is dynamic and is revised frequently as new data become available. New species are added to the list, and others are dropped from the list as appropriate. -
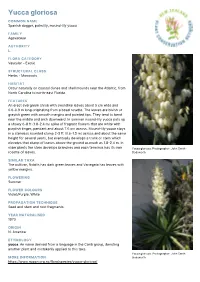
Yucca Gloriosa
Yucca gloriosa COMMON NAME Spanish dagger, palm lily, mound-lily yucca FAMILY Agavaceae AUTHORITY L. FLORA CATEGORY Vascular – Exotic STRUCTURAL CLASS Herbs - Monocots HABITAT Occur naturally on coastal dunes and shell mounds near the Atlantic, from North Carolina to north-east Florida. FEATURES An erect evergreen shrub with swordlike leaves about 5 cm wide and 0.6-0.9 m long originating from a basal rosette. The leaves are bluish or grayish green with smooth margins and pointed tips. They tend to bend near the middle and arch downward. In summer mound-lily yucca puts up a showy 6-8 ft (1.8-2.4 m) spike of fragrant flowers that are white with purplish tinges, pendant and about 7.6 cm across. Mound-lily yucca stays in a stemless rounded clump 2-5 ft (0.6-1.5 m) across and about the same height for several years, but eventually develops a trunk or stem which elevates that clump of leaves above the ground as much as 1.8-2.4 m. In older plants the stem develops branches and each terminus has its own Yucca gloriosa. Photographer: John Smith- rosette of leaves. Dodsworth SIMILAR TAXA The cultivar, Nobilis has dark green leaves and Variegata has leaves with yellow margins. FLOWERING Summer FLOWER COLOURS Violet/Purple, White PROPAGATION TECHNIQUE Seed and stem and root fragments. YEAR NATURALISED 1970 ORIGIN N. America ETYMOLOGY yucca: An name derived from a language in the Carib group, denoting another plant and mistakenly applied to this taxa. Yucca gloriosa. Photographer: John Smith- MORE INFORMATION Dodsworth https://www.nzpcn.org.nz/flora/species/yucca-gloriosa/. -

Transcript Profiling of a Novel Plant Meristem, the Monocot Cambium
Journal of Integrative JIPB Plant Biology Transcript profiling of a novel plant meristem, the monocot cambiumFA Matthew Zinkgraf1,2, Suzanne Gerttula1 and Andrew Groover1,3* 1. US Forest Service, Pacific Southwest Research Station, Davis, California, USA 2. Department of Computer Science, University of California, Davis, USA 3. Department of Plant Biology, University of California, Davis, USA Article *Correspondence: Andrew Groover ([email protected]) doi: 10.1111/jipb.12538 Abstract While monocots lack the ability to produce a xylem tissues of two forest tree species, Populus Research vascular cambium or woody growth, some monocot trichocarpa and Eucalyptus grandis. Monocot cambium lineages evolved a novel lateral meristem, the monocot transcript levels showed that there are extensive overlaps cambium, which supports secondary radial growth of between the regulation of monocot cambia and vascular stems. In contrast to the vascular cambium found in woody cambia. Candidate regulatory genes that vary between the angiosperm and gymnosperm species, the monocot monocot and vascular cambia were also identified, and cambium produces secondary vascular bundles, which included members of the KANADI and CLE families involved have an amphivasal organization of tracheids encircling a in polarity and cell-cell signaling, respectively. We suggest central strand of phloem. Currently there is no information that the monocot cambium may have evolved in part concerning the molecular genetic basis of the develop- through reactivation of genetic mechanisms involved in ment or evolution of the monocot cambium. Here we vascular cambium regulation. report high-quality transcriptomes for monocot cambium Edited by: Chun-Ming Liu, Institute of Crop Science, CAAS, China and early derivative tissues in two monocot genera, Yucca Received Feb. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -

Complete Chloroplast Genomes Shed Light on Phylogenetic
www.nature.com/scientificreports OPEN Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae) Ju Namgung1,4, Hoang Dang Khoa Do1,2,4, Changkyun Kim1, Hyeok Jae Choi3 & Joo‑Hwan Kim1* Allioideae includes economically important bulb crops such as garlic, onion, leeks, and some ornamental plants in Amaryllidaceae. Here, we reported the complete chloroplast genome (cpDNA) sequences of 17 species of Allioideae, fve of Amaryllidoideae, and one of Agapanthoideae. These cpDNA sequences represent 80 protein‑coding, 30 tRNA, and four rRNA genes, and range from 151,808 to 159,998 bp in length. Loss and pseudogenization of multiple genes (i.e., rps2, infA, and rpl22) appear to have occurred multiple times during the evolution of Alloideae. Additionally, eight mutation hotspots, including rps15-ycf1, rps16-trnQ-UUG, petG-trnW-CCA , psbA upstream, rpl32- trnL-UAG , ycf1, rpl22, matK, and ndhF, were identifed in the studied Allium species. Additionally, we present the frst phylogenomic analysis among the four tribes of Allioideae based on 74 cpDNA coding regions of 21 species of Allioideae, fve species of Amaryllidoideae, one species of Agapanthoideae, and fve species representing selected members of Asparagales. Our molecular phylogenomic results strongly support the monophyly of Allioideae, which is sister to Amaryllioideae. Within Allioideae, Tulbaghieae was sister to Gilliesieae‑Leucocoryneae whereas Allieae was sister to the clade of Tulbaghieae‑ Gilliesieae‑Leucocoryneae. Molecular dating analyses revealed the crown age of Allioideae in the Eocene (40.1 mya) followed by diferentiation of Allieae in the early Miocene (21.3 mya). The split of Gilliesieae from Leucocoryneae was estimated at 16.5 mya. -

Dyuhei Sato Division of Genetics, Bot. Inst. Faculty of Science, Tokyo
ANALYSIS OF THE KARYOTYPES IN YUCCA, A GA VE AND THE RELATED GENERA WITH SPECIAL REFERENCE TO THE PHYLOGENETIC SIGNIFICANCEI~ Dyuhei SATo Divisionof Genetics, Bot. Inst. Faculty of Science, Tokyo Imperial University McKelvey and Sax (2933) have called attention to the existence of taxonomic and cytological similarities of the genera Yucca, Hesperoyucca, Gleistvucca,Hesperoaloe and Samuela of the Liliaceae with the genera Agave and Fourcroya which belong to a related family, Amaryllidaceae. Wh.itaker (1934) also has reported that Polianhes and Fourcroya have exactly the same chromosome constitution as the Yucca-Abave karyotype (5 long and 25 short chromosomes) (Figs. 1, 2). These observations when considered in respect to taxonomic resemblances, seem to indicate that the genera mentioned above are more closely related than it is shown by their classifica- tion into distinct families. Whitaker also has remarked that Dasylirion (2n=38) and ATolina(2n=36) in Yucceae and Doryanthes (2n=36) in Agavoideae are of different karyotypes from the Yucca-Agave type. In the present work an analysis of the karyotypes in Liliaceous plants has been attempted and several karyotypes have been found in Scilloideae. Eucornis and Carassia have been selected with the purpose of discovering a possible connecting link between these genera and the Yucca-Agave group. In the present paper an analysis of the karyotypes of the following species is given. LILIACEAE Scilloideae 211 Fig. Euconis undulata 60=8L+8M+44S (4b)2) 3 Euconsispallidi ora 60=8L+8M+44S (4b) 4 Eucomispunctata 60=8L±8M+44S (4b) 5 Camassiaescrema 30=6L+24S (2b) 6 Yucceae Yuccafilamentosa 30 60=1OL+50S (2b) 1, 7 Yuccarecurvifolia 30 60=1OL+50S (2b) 2, 8 Yuccaaloifolia 60=1OL+50S (2b) 9 „ var. -

Agave Americana and Furcraea Andina: Key Species to Andean Cultures in Ecuador
Ethnobotany Agave americana and Furcraea andina: Key Species to Andean Cultures in Ecuador LUCÍA DE LA TORRE1*, IAN CUMMINS2, AND ELIOT LOGAN-HINES2 Botanical Sciences 96 (2): 246-266, 2018 Abstract Background: The rich Agaveae-based culture that exists in the Ecuadorian Andes is little known. Wild DOI: 10.17129/botsci.1813 and cultivated rosettes of Agave americana and Furcraea andina coexist in arid Andean landscapes. A. americana is considered an introduced species to Ecuador. Received: Questions: What are Agaveae use patterns and cultural importance in the Ecuadorian Andes? Is the ethno- December 19th, 2017 Accepted: botanical significance of Agave in Ecuador comparable to that in Mexico and other Andean countries? Agave americana, Furcraea andina March 12th, 2018 Species studied: Associated editor: Study site, dates: Ecuadorian Andes, 2016. Salvadro Arias Methods: Semi-structured interviews to Agaveae users (37) and a review of literature on ethnobotanical research conducted in Ecuador since the 18th century. Results: A. americana is more diversely and widely used than F. andina (124 vs 36 uses and 548 vs 140 use records, respectively). The versatility of A. americana lies in its mishki (sap extracted from its heart) which has multiple medicinal, edible and ceremonial applications. We found significant variation of its use patterns throughout the region. The main use of F. andina as a source of fiber is disappearing. Most productive initiatives involve A. americana (92 %, n = 53). Conclusion: The importance of A. americana in the Ecuadorian Andes is comparable to that of agaves in Mexico, but not to its importance in other Andean countries where it is used sporadically. -

ISB: Atlas of Florida Vascular Plants
Longleaf Pine Preserve Plant List Acanthaceae Asteraceae Wild Petunia Ruellia caroliniensis White Aster Aster sp. Saltbush Baccharis halimifolia Adoxaceae Begger-ticks Bidens mitis Walter's Viburnum Viburnum obovatum Deer Tongue Carphephorus paniculatus Pineland Daisy Chaptalia tomentosa Alismataceae Goldenaster Chrysopsis gossypina Duck Potato Sagittaria latifolia Cow Thistle Cirsium horridulum Tickseed Coreopsis leavenworthii Altingiaceae Elephant's foot Elephantopus elatus Sweetgum Liquidambar styraciflua Oakleaf Fleabane Erigeron foliosus var. foliosus Fleabane Erigeron sp. Amaryllidaceae Prairie Fleabane Erigeron strigosus Simpson's rain lily Zephyranthes simpsonii Fleabane Erigeron vernus Dog Fennel Eupatorium capillifolium Anacardiaceae Dog Fennel Eupatorium compositifolium Winged Sumac Rhus copallinum Dog Fennel Eupatorium spp. Poison Ivy Toxicodendron radicans Slender Flattop Goldenrod Euthamia caroliniana Flat-topped goldenrod Euthamia minor Annonaceae Cudweed Gamochaeta antillana Flag Pawpaw Asimina obovata Sneezeweed Helenium pinnatifidum Dwarf Pawpaw Asimina pygmea Blazing Star Liatris sp. Pawpaw Asimina reticulata Roserush Lygodesmia aphylla Rugel's pawpaw Deeringothamnus rugelii Hempweed Mikania cordifolia White Topped Aster Oclemena reticulata Apiaceae Goldenaster Pityopsis graminifolia Button Rattlesnake Master Eryngium yuccifolium Rosy Camphorweed Pluchea rosea Dollarweed Hydrocotyle sp. Pluchea Pluchea spp. Mock Bishopweed Ptilimnium capillaceum Rabbit Tobacco Pseudognaphalium obtusifolium Blackroot Pterocaulon virgatum -

Low-Maintenance Landscape Plants for South Florida1
ENH854 Low-Maintenance Landscape Plants for South Florida1 Jody Haynes, John McLaughlin, Laura Vasquez, Adrian Hunsberger2 Introduction regular watering, pruning, or spraying—to remain healthy and to maintain an acceptable aesthetic This publication was developed in response to quality. A low-maintenance plant has low fertilizer requests from participants in the Florida Yards & requirements and few pest and disease problems. In Neighborhoods (FYN) program in Miami-Dade addition, low-maintenance plants suitable for south County for a list of recommended landscape plants Florida must also be adapted to—or at least suitable for south Florida. The resulting list includes tolerate—our poor, alkaline, sand- or limestone-based over 350 low-maintenance plants. The following soils. information is included for each species: common name, scientific name, maximum size, growth rate An additional criterion for the plants on this list (vines only), light preference, salt tolerance, and was that they are not listed as being invasive by the other useful characteristics. Florida Exotic Pest Plant Council (FLEPPC, 2001), or restricted by any federal, state, or local laws Criteria (Burks, 2000). Miami-Dade County does have restrictions for planting certain species within 500 This section will describe the criteria by which feet of native habitats they are known to invade plants were selected. It is important to note, first, that (Miami-Dade County, 2001); caution statements are even the most drought-tolerant plants require provided for these species. watering during the establishment period. Although this period varies among species and site conditions, Both native and non-native species are included some general rules for container-grown plants have herein, with native plants denoted by †. -

Appendixes, Comprehensive Conservation Plan, Rainwater
Glossary accessible—Pertaining to physical access to areas and biological control—Use of organisms or viruses to activities for people of different abilities, especially control invasive plants or other pests. those with physical impairments. biological diversity, also biodiversity—Variety of life adaptive resource management—Rigorous application and its processes, including the variety of living of management, research, and monitoring to gain organisms, the genetic differences among them, and information and experience necessary to assess and the communities and ecosystems in which they occur modify management activities; a process that uses (The Fish and Wildlife Service Manual, 052 FW 1.12B). feedback from research, monitoring, and evaluation of The National Wildlife Refuge System’s focus is on management actions to support or modify objectives indigenous species, biotic communities, and ecological and strategies at all planning levels; a process in processes. which policy decisions are carried out within a framework of scientifi cally driven experiments to biotic—Pertaining to life or living organisms; caused, test predictions and assumptions inherent in a produced by, or comprising living organisms. management plan. Analysis of results helps managers CAFO—Concentrated animal-feeding operation. determine whether current management should continue as is or whether it should be modifi ed to canopy—Layer of foliage, generally the uppermost achieve desired conditions. layer, in a vegetative stand; midlevel or understory vegetation in multilayered stands; canopy closure Administration Act—National Wildlife Refuge System (also canopy cover) is an estimate of the amount of Administration Act of 1966. overhead vegetative cover. alternative —Reasonable way to solve an identifi ed catabolized (catabolism)—Breakdown of more complex problem or satisfy the stated need (40 CFR 1500.2); substances into simpler ones, with the release of one of several different means of accomplishing refuge energy. -

D.1A Floral and Faunal Compendia
Appendix D.1a Floral and Faunal Compendia APPENDIX D.1a FLORAL AND FAUNAL COMPENDIA INTRODUCTION TO FLORAL AND FAUNAL SURVEY Expected site use by wildlife is derived from survey information combined with documented habitat preferences of regional wildlife species, which, whether or not recorded during the survey, are considered likely to include the project area within their range. Habitat designations used in this report are according to the classification system of Holland (1986). Floral taxonomy used in this report follows the Jepson Manual (Hickman 1993), with updates in accordance to the online Jepson Interchange where known. Common plant names, where not available from Munz (1974), are taken from Abrams (1923), Robbins, et al. (1951), Collins (1972), Niehaus and Ripper (1976) and Muns (1983). Vertebrates identified in the field by sight, calls, tracks, scat or other signs are cited according to the nomenclature of Jennings (1983) for amphibians and reptiles; AOU (1983) for birds; and Jones, et al. (1982) for mammals. Butterflies observed or collected in the field were identified with the help of Garth and Tilden (1986) and Tilden and Smith (1986). FLORAL COMPENDIUM1 LEGEND * Nonnative @ Ornamental/Landscape VASCULAR PLANTS CONIFERAE PINACEAE - PINE FAMILY * Pinus halepensis Aleppo Pine ANGIOSPERMAE (DICOTYLEDONS) ANACARDIACEAE - SUMAC FAMILY Malosma laurina laurel sumac Rhus ovata sugar bush * Schinus molle Peruvian pepper-tree Toxicodendron diversilobum poison-oak APOCYNACEAE - DOGBANE FAMILY * Vinca major periwinkle * Nerium oleander oleander ASTERACEAE - SUNFLOWER FAMILY Baccharis pilularis coyote brush Baccharis salicifolia mulefat Helianthus gracilentus slender sunflower * Picris echioides bristly ox-tongue * Silybum marianum milk thistle * Sonchus asper prickly sow-thistle * Sonchus oleraceus common sow-thistle Stephanomeria virgata twiggy wreathplant BRASSICACEAE - MUSTARD FAMILY * Brassica nigra black mustard * Sisymbrium officinale hedge-mustard CAPRIFOLIACEAE - HONEYSUCKLE FAMILY Sambuccus. -

Lush & Efficient
R EVISED E DITION LLushush && EEfficientfficient LandscapeLandscape GardeningGardening inin thethe CoachellaCoachella ValleyValley Coachella Valley Water District LLushush && EEfficientfficient LandscapeLandscape GardeningGardening inin thethe CoachellaCoachella ValleyValley RR EVISEDEVISED EE DITIONDITION CoachellaCoachella ValleyValley WaterWater DistrictDistrict IRONWOOD PRESS Tucson, Arizona Coachella Valley Cover photo by Acknowledgements A special thank you goes to Water District Scott Millard Directors and staff of the Ann Copeland, now retired Coachella Valley Water District, Primary photography by Coachella Valley Water District from CVWD. An educational specialist who taught water CVWD, is a local govern- Scott Millard: © pages 5, 7, 8, 9, extend their gratitude to Scott ment agency controlled by Millard of Ironwood Press science to the children of five directors elected by the 10 (right), 11, 13, 14, 15, 17, 19, Coachella Valley, she took on 20, 21, 22 (left), 23, 24, 25, 26, in Tucson, Ariz., for bringing registered voters within its this revised book to fruition. the additional responsibility 1,000 square mile service area. 27, 28, 35, 38, 41, 42, 43, 44, 45 of working closely with Eric That area in the southeastern (top & lower right), 46 (top left, Scott and primary author Eric A. Johnson were partners at Johnson, reading his text and California desert extends from bottom center & bottom right), identifying photos to illustrate west of Palm Springs to the 47 (bottom left inset, bottom Ironwood Press and published communities along the Salton several excellent desert land- it. She also worked closely right & upper right), 48 (left & with contributing author Dave Sea. It is located primarily in upper left), 49, 50, 51, 52, 53, 54, scaping books together before Riverside County but extends Harbison in developing and 55 (left & center inset & right), Eric’s death.