Fosphenytoin Sodium Injection, USP Rx Only WARNING
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Optum Essential Health Benefits Enhanced Formulary PDL January
PENICILLINS ketorolac tromethamineQL GENERIC mefenamic acid amoxicillin/clavulanate potassium nabumetone amoxicillin/clavulanate potassium ER naproxen January 2016 ampicillin naproxen sodium ampicillin sodium naproxen sodium CR ESSENTIAL HEALTH BENEFITS ampicillin-sulbactam naproxen sodium ER ENHANCED PREFERRED DRUG LIST nafcillin sodium naproxen DR The Optum Preferred Drug List is a guide identifying oxacillin sodium oxaprozin preferred brand-name medicines within select penicillin G potassium piroxicam therapeutic categories. The Preferred Drug List may piperacillin sodium/ tazobactam sulindac not include all drugs covered by your prescription sodium tolmetin sodium drug benefit. Generic medicines are available within many of the therapeutic categories listed, in addition piperacillin sodium/tazobactam Fenoprofen Calcium sodium to categories not listed, and should be considered Meclofenamate Sodium piperacillin/tazobactam as the first line of prescribing. Tolmetin Sodium Amoxicillin/Clavulanate Potassium LOW COST GENERIC PREFERRED For benefit coverage or restrictions please check indomethacin your benefit plan document(s). This listing is revised Augmentin meloxicam periodically as new drugs and new prescribing LOW COST GENERIC naproxen kit information becomes available. It is recommended amoxicillin that you bring this list of medications when you or a dicloxacillin sodium CARDIOVASCULAR covered family member sees a physician or other penicillin v potassium ACE-INHIBITORS healthcare provider. GENERIC QUINOLONES captopril ANTI-INFECTIVES -

Supplementary Materials
Supplementary Materials Table S1. The significant drug pairs in potential DDIs examined by the two databases. Micromedex Drugs.com List of drugs paired PK-PD Mechanism details 1. Amiodarone— PD Additive QT-interval prolongation Dronedarone 2. Amiodarone— PK CYP3A inhibition by Ketoconazole Ketoconazole 3. Ciprofloxacin— PD Additive QT-interval prolongation Dronedarone 4. Cyclosporine— PK CYP3A inhibition by Cyclosporine Dronedarone 5. Dronedarone— PK CYP3A inhibition by Erythromycin Erythromycin 6. Dronedarone— PD Additive QT-interval prolongation Flecainide 7. Dronedarone— PK CYP3A4 inhibition by Itraconazole Itraconazole 8. Dronedarone— PK Contraindication Major CYP3A inhibition by Ketoconazole Ketoconazole 9. Dronedarone— PD Additive QT-interval prolongation Procainamide PD 10. Dronedarone—Sotalol Additive QT-interval prolongation 11. Felodipine— PK CYP3A inhibition by Itraconazole Itraconazole 12. Felodipine— PK CYP3A inhibition by Ketoconazole Ketoconazole 13. Itraconazole— PK CYP3A inhibition by Itraconazole Nisoldipine 14. Ketoconazole— PK CYP3A inhibition by Ketoconazole Nisoldipine 15. Praziquantel— PK CYP induction by Rifampin Rifampin PD 1. Amikacin—Furosemide Additive or synergistic toxicity 2. Aminophylline— Decreased clearance of PK Ciprofloxacin Theophylline by Ciprofloxacin 3. Aminophylline— PK Decreased hepatic metabolism Mexiletine 4. Amiodarone— PD Additive effects on QT interval Ciprofloxacin 5. Amiodarone—Digoxin PK P-glycoprotein inhibition by Amiodarone 6. Amiodarone— PD, PK Major Major Additive effects on QT Erythromycin prolongation, CYP3A inhibition by Erythromycin 7. Amiodarone— PD, PK Flecainide Antiarrhythmic inhibition by Amiodarone, CYP2D inhibition by Amiodarone 8. Amiodarone— PK CYP3A inhibition by Itraconazole Itraconazole 9. Amiodarone— PD Antiarrhythmic inhibition by Procainamide Amiodarone 10. Amiodarone— PK CYP induction by Rifampin Rifampin PD Additive effects on refractory 11. Amiodarone—Sotalol potential 12. Amiodarone— PK CYP3A inhibition by Verapamil Verapamil 13. -

Grapefruit Juice May Cause Prescription Drug Interactions
Grapefruit Juice May Cause Prescription Drug Interactions Grapefruit juice provides many nutrients, including vitamin C, potassium and lycopene. But chemicals in grapefruit juice and grapefruit pulp interfere with the enzymes that break down (metabolize) various drugs in the digestive system — including certain calcium channel blockers and cholesterol- lowering drugs. The result can be excessively high levels of these drugs in the blood and an increased risk of potentially serious side effects. Pomelos and Seville oranges, a type of bitter orange often used to make marmalade and compotes, may have a similar effect. Here's a sampling of drugs known to have potentially serious interactions with grapefruit products: Drug name Type of drug Amiodarone (Cordarone) A drug used to treat and prevent abnormal heart rhythms (arrhythmias) Buspirone (BuSpar), sertraline (Zoloft) Antidepressants Carbamazepine (Carbatrol, Tegretol) An anti-seizure medication Cyclosporine (Neoral, Sandimmune), tacrolimus Immunosuppressant drugs (Prograf) Felodipine (Plendil), nifedipine (Procardia), Calcium channel blockers used to treat high blood nimodipine (Nimotop), nisoldipine (Sular) pressure Saquinavir An HIV medication Simvastatin (Zocor), lovastatin (Mevacor), Statins used to treat high cholesterol atorvastatin (Lipitor) If you're concerned about the effect grapefruit juice may have on your medications, talk to your doctor or pharmacist. In some cases, it may be important to avoid grapefruit and grapefruit products, as well as pomelos, Seville oranges and products made with these fruits. Waiting to take these medications — even up to 24 hours — after you drink grapefruit juice won't prevent an interaction. In other cases, it may be possible to switch to an alternative medication that won't interact with these fruits. -

Download Product Insert (PDF)
PRODUCT INFORMATION Nisoldipine Item No. 20998 CAS Registry No.: 63675-72-9 Formal Name: 1,4-dihydro-2,6-dimethyl-4-(2- nitrophenyl)-3,5-pyridinedicarboxylic acid, 3-methyl 5-(2-methylpropyl) ester NO Synonyms: (±)-BAY-K-5552, (±)-Nisoldipine 2 O O MF: C20H24N2O6 FW: 388.4 O O Purity: ≥97% UV/Vis.: λ: 235, 330 nm max N Supplied as: A crystalline solid Storage: Room temperature H Stability: ≥2 years Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Nisoldipine is supplied as a crystalline solid. A stock solution may be made by dissolving the nisoldipine in the solvent of choice. Nisoldipine is soluble in organic solvents such as ethanol, DMSO, and dimethyl formamide (DMF), which should be purged with an inert gas. The solubility of nisoldipine in ethanol is approximately 3 mg/ml and approximately 30 mg/ml in DMSO and DMF. Nisoldipine is sparingly soluble in aqueous buffers. For maximum solubility in aqueous buffers, nisoldipine should first be dissolved in DMSO and then diluted with the aqueous buffer of choice. Nisoldipine has a solubility of approximately 0.1 mg/ml in a 1:10 solution of DMSO:PBS (pH 7.2) using this method. We do not recommend storing the aqueous solution for more than one day. Description Nisoldipine is a calcium channel inhibitor.1 It binds to calcium channels in isolated rat ventricular 1,2 membranes (Kd = 0.04 nM) and inhibits calcium uptake by smooth muscle cells. Nisoldipine inhibits 1 acetylcholine-induced contraction of isolated rabbit coronary arteries (IC50 = 0.03 nM). -

2021 Formulary List of Covered Prescription Drugs
2021 Formulary List of covered prescription drugs This drug list applies to all Individual HMO products and the following Small Group HMO products: Sharp Platinum 90 Performance HMO, Sharp Platinum 90 Performance HMO AI-AN, Sharp Platinum 90 Premier HMO, Sharp Platinum 90 Premier HMO AI-AN, Sharp Gold 80 Performance HMO, Sharp Gold 80 Performance HMO AI-AN, Sharp Gold 80 Premier HMO, Sharp Gold 80 Premier HMO AI-AN, Sharp Silver 70 Performance HMO, Sharp Silver 70 Performance HMO AI-AN, Sharp Silver 70 Premier HMO, Sharp Silver 70 Premier HMO AI-AN, Sharp Silver 73 Performance HMO, Sharp Silver 73 Premier HMO, Sharp Silver 87 Performance HMO, Sharp Silver 87 Premier HMO, Sharp Silver 94 Performance HMO, Sharp Silver 94 Premier HMO, Sharp Bronze 60 Performance HMO, Sharp Bronze 60 Performance HMO AI-AN, Sharp Bronze 60 Premier HDHP HMO, Sharp Bronze 60 Premier HDHP HMO AI-AN, Sharp Minimum Coverage Performance HMO, Sharp $0 Cost Share Performance HMO AI-AN, Sharp $0 Cost Share Premier HMO AI-AN, Sharp Silver 70 Off Exchange Performance HMO, Sharp Silver 70 Off Exchange Premier HMO, Sharp Performance Platinum 90 HMO 0/15 + Child Dental, Sharp Premier Platinum 90 HMO 0/20 + Child Dental, Sharp Performance Gold 80 HMO 350 /25 + Child Dental, Sharp Premier Gold 80 HMO 250/35 + Child Dental, Sharp Performance Silver 70 HMO 2250/50 + Child Dental, Sharp Premier Silver 70 HMO 2250/55 + Child Dental, Sharp Premier Silver 70 HDHP HMO 2500/20% + Child Dental, Sharp Performance Bronze 60 HMO 6300/65 + Child Dental, Sharp Premier Bronze 60 HDHP HMO -
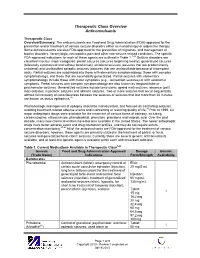
Therapeutic Class Overview Anticonvulsants
Therapeutic Class Overview Anticonvulsants Therapeutic Class Overview/Summary: The anticonvulsants are Food and Drug Administration (FDA)-approved for the prevention and/or treatment of various seizure disorders either as monotherapy or adjunctive therapy. Some anticonvulsants are also FDA-approved for the prevention of migraines, and management of bipolar disorders, fibromyalgia, neuropathic pain and other non-seizure related conditions. The specific FDA-approved indications for each of these agents are outlined in Table 1.1-44 Seizure disorders are classified into four major categories: partial seizures (seizures beginning locally), generalized seizures (bilaterally symmetrical and without local onset), unilateral seizures (seizures that are predominantly unilateral) and unclassified epileptic seizures (seizures that are unclassifiable because of incomplete data). Partial seizures are subdivided into those with elementary symptomatology, those with complex symptomatology, and those that are secondarily generalized. Partial seizures with elementary symptomatology include those with motor symptoms (e.g., Jacksonian seizures) or with autonomic symptoms. Partial seizures with complex symptomatology are also known as temporal lobe or psychomotor seizures. Generalized seizures include tonic-clonic (grand mal) seizures, absence (petit mal) seizures, myoclonic seizures and akinetic seizures. Two or more seizures that occur sequentially without full recovery of consciousness between the seizures or seizures that last more than 30 minutes are known as status epilepticus.45 Pharmacologic management of epilepsy should be individualized, and focused on controlling seizures, avoiding treatment-related adverse events and maintaining or restoring quality of life.46 Prior to 1990, six major antiepileptic drugs were available for the treatment of various forms of epilepsy, including carbamazepine, ethosuximide, phenobarbital, phenytoin, primidone and valproic acid. -

Calcium Channel Blockers
Calcium Channel Blockers Summary In general, calcium channel blockers (CCBs) are used most often for the management of hypertension and angina. There are 2 classes of CCBs: the dihydropyridines (DHPs), which have greater selectivity for vascular smooth muscle cells than for cardiac myocytes, and the non-DHPs, which have greater selectivity for cardiac myocytes and are used for cardiac arrhythmias. The DHPs cause peripheral edema, headaches, and postural hypotension most commonly, all of which are due to the peripheral vasodilatory effects of the drugs in this class of CCBs. The non-DHPs are negative inotropes and chronotropes; they can cause bradycardia and depress AV node conduction, increasing the risk of heart failure exacerbation, bradycardia, and AV block. Clevidipine is a DHP calcium channel blocker administered via continuous IV infusion and used for rapid blood pressure reductions. All CCBs are substrates of CYP3A4, but both diltiazem and verapamil are also inhibitors of 3A4 and have an increased risk of drug interactions. Verapamil also inhibits CYP2C9, CYP2C19, and CYP1A2. Pharmacology CCBs selectively inhibit the voltage-gated L-type calcium channels on cardiac myocytes, vascular smooth muscle cells, and cells within the sinoatrial (SA) and atrioventricular (AV) nodes, preventing influx of extracellular calcium. CCBs act by either deforming the channels, inhibiting ion-control gating mechanisms, and/or interfering with the release of calcium from the major cellular calcium store, the endoplasmic reticulum. Calcium influx via these channels serves for excitation-contraction coupling and electrical discharge in the heart and vasculature. A decrease in intracellular calcium will result in inhibition of the contractile process of the myocardial smooth muscle cells, resulting in dilation of the coronary and peripheral arterial vasculature. -

Clinically Relevant Drug Interactions with Antiepileptic Drugs
British Journal of Clinical Pharmacology DOI:10.1111/j.1365-2125.2005.02529.x Clinically relevant drug interactions with antiepileptic drugs Emilio Perucca Institute of Neurology IRCCS C. Mondino Foundation, Pavia, and Clinical Pharmacology Unit, Department of Internal Medicine and Therapeutics, University of Pavia, Pavia, Italy Correspondence Some patients with difficult-to-treat epilepsy benefit from combination therapy with Emilio Perucca MD, PhD, Clinical two or more antiepileptic drugs (AEDs). Additionally, virtually all epilepsy patients will Pharmacology Unit, Department of receive, at some time in their lives, other medications for the management of Internal Medicine and Therapeutics, associated conditions. In these situations, clinically important drug interactions may University of Pavia, Piazza Botta 10, occur. Carbamazepine, phenytoin, phenobarbital and primidone induce many cyto- 27100 Pavia, Italy. chrome P450 (CYP) and glucuronyl transferase (GT) enzymes, and can reduce Tel: + 390 3 8298 6360 drastically the serum concentration of associated drugs which are substrates of the Fax: + 390 3 8222 741 same enzymes. Examples of agents whose serum levels are decreased markedly by E-mail: [email protected] enzyme-inducing AEDs, include lamotrigine, tiagabine, several steroidal drugs, cyclosporin A, oral anticoagulants and many cardiovascular, antineoplastic and psy- chotropic drugs. Valproic acid is not enzyme inducer, but it may cause clinically relevant drug interactions by inhibiting the metabolism of selected substrates, most Keywords notably phenobarbital and lamotrigine. Compared with older generation agents, most antiepileptic drugs, drug interactions, of the recently developed AEDs are less likely to induce or inhibit the activity of CYP enzyme induction, enzyme inhibition, or GT enzymes. However, they may be a target for metabolically mediated drug epilepsy, review interactions, and oxcarbazepine, lamotrigine, felbamate and, at high dosages, topira- mate may stimulate the metabolism of oral contraceptive steroids. -

Nisoldipine (Sular)
ALDERFER & TRAVIS CARDIOLOGY, PC 670 Lawn Ave., Suite 3A Sellersville, PA 18960 Tel (215)257-9500 FAX (215) 257-9500 NISOLDIPINE SUSTAINED ACTION - ORAL OTHER NAMES: Sular USES: This drug is a calcium channel blocker. Calcium is involved in muscle contraction. By blocking calcium, this medication relaxes the heart muscle and dilates the blood vessels which lowers blood pressure. This drug is used to treat hypertension (high blood pressure). HOW TO TAKE THIS MEDICATION: May be taken with or without food usually once daily. Do not take with a high fat meal. Do not chew, divide or crush. Tablets should be swallowed whole preferably with a full glass of water. Do not stop taking suddenly. SIDE EFFECTS: This drug may cause dizziness and lightheadedness especially during the first few days. Avoid activities requiring alertness. When you sit or lie down for a while, get up slowly to minimize dizziness and allow your body to adjust. You may also experience bloating, heartburn, nausea, blurred vision, muscle cramps, headache, flushing, sweating and sleep disturbances. These effects should disappear as your body adjusts to the medication. Inform your doctor if they become bothersome. Notify your doctor if you develop breathing difficulties, rash, swelling of the hands or feet, weight gain, chest pain or an irregular heartbeat. PRECAUTIONS: Before having surgery, including dental surgery, tell the doctor that you take this drug. Tell your doctor of any diseases you have especially liver disease or if you've had congestive heart failure or if you have any allergies. Limit using alcohol while taking this medication. -

Development of Fast Dissolving Tablets of Nisoldipine by Solid Dispersion Technology Using Poloxamer 407 and Poloxamer 188
J Young Pharm, 2016; 8(4): 341-349 A multifaceted peer reviewed journal in the field of Pharmacy Original Article www.jyoungpharm.org | www.phcog.net Development of Fast Dissolving Tablets of Nisoldipine by Solid Dispersion Technology using Poloxamer 407 and Poloxamer 188 Manimaran Vasanthan and Damodharan Narayanasamy* Department of Pharmaceutics, SRM College of Pharmacy, SRM University, Kattankulathur, Tamilnadu, INDIA. ABSTRACT Objective: The aim of the present study is to design oral fast-release tab- and its formulation with different carriers used in the preparation of solid lets of nisoldipine and to optimize the drug dissolution profile by modifying dispersions. X-ray diffraction studies revealed that the degree of crystallin- the carrier concentration. Poloxamer 407 and Poloxamer 188 were selected ity of nisoldipine decreased when the concentration of carriers increased, as carriers for preparing the solid dispersion (SD) by solvent evaporation which showed that the drug is in amorphous nature. method with different drug polymer ratios. Methods: The prepared solid dispersions were analyzed for physical state, drug:carrier interactions by X- Key words: Fast dissolving tablets, Poloxamer 407, Poloxamer 188, Solid ray diffraction, infrared spectroscopy, differential scanning calorimetry and dispersion, Superdisintergrants. scanning electron microscopy. Results: The dissolution studies revealed that all solid dispersions showed increased dissolution rate whencompared Correspondence : to pure nisoldipine. Among the two polymers used, poloxamer 407(P 407) Prof. N. Damodharan, was found to be better than poloxamer 188(P 188) in the enhancement of Professor and Head, Department of Pharmaceutics, SRM College of Pharmacy, dissolution efficiency. The tablets were formulated using solid dispersion SRM University, Kattankulathur, Kancheepuram, INDIA. -

Treatment in Hypertension Inadequately Controlled by Atenolol B.I
Postgrad Med J (1993) 69, 117- 120 C The Fellowship of Postgraduate Medicine, 1993 Postgrad Med J: first published as 10.1136/pgmj.69.808.117 on 1 February 1993. Downloaded from Comparison ofnisoldipine and nifedipine as additional treatment in hypertension inadequately controlled by atenolol B.I. Hoffbrand, K.A. Earle, J.G. Nievell, L.J. Restrick and N.J. Simmonds Department ofMedicine, Whittington Hospital, London, N19 SNFand 'Hypertension Clinic, Royal Northern Hospital, London, N7 6LD, UK Summary: Twenty-eight patients (11 Caucasian, 17 black) whose blood pressure was more than 160/96 mmHg after 4 weeks on placebo added to atenolol 100 mg/day were randomly given, in addition, nisoldipine 10 mg or nifedipine 20 mg each twice a day for 8 weeks in a double-blind cross-over study. There was a statistically significant (P < 0.001) fall in blood pressure with no change in heart rate, both supine and erect, on both drugs. There were no significant differences between nisoldipine and nifedipine. Adverse effects were recorded in 15%, 17% and 35% of the patients available for safety comparison for placebo, nisoldipine and nifedipine, respectively. There were no significant differences between the black and Caucasian patients in blood pressure responses, although the study had only a low power to detect these. However, the fasting serum triglyceride levels at the end of both calcium antagonist treatment periods were highly significantly lower in the black patients compared with the Caucausian patients. Nisoldipine, which has a higher coronary vascular selectivity and less negative inotropism than nifedipine, Protected by copyright. is as effective and as well tolerated as nifedipine in patients whose hypertension is inadequately controlled on atenolol. -

Principal Component Analysis of HPLC Retention Data and Molecular Modeling Structural Parameters of Cardiovascular System Drugs
Int. J. Mol. Sci. 2010, 11, 2681-2698; doi:10.3390/ijms11072681 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Principal Component Analysis of HPLC Retention Data and Molecular Modeling Structural Parameters of Cardiovascular System Drugs in View of Their Pharmacological Activity Jolanta Stasiak 1, Marcin Koba 1, Leszek Bober 2 and Tomasz Bączek 3,* 1 Department of Medicinal Chemistry, Faculty of Pharmacy, Collegium Medicum, Nicolaus Copernicus University, Bydgoszcz, Poland; E-Mails: [email protected] (J.S.); [email protected] (M.K.) 2 Polpharma SA Pharmaceutical Works, Starogard Gdański, Poland; E-Mail: [email protected] 3 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Medical University of Gdańsk, Gdańsk, Poland * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +48-58-349-31-35; Fax: +48-58-349-31-30. Received: 22 April 2010; in revised form: 11 June 2010 / Accepted: 28 June 2010 / Published: 9 July 2010 Abstract: Evaluation of relationships between molecular modeling structural parameters and high-performance liquid chromatography (HPLC) retention data of 11 cardiovascular system drugs by principal component analysis (PCA) in relation to their pharmacological activity was performed. The six retention data parameters were determined on three different HPLC columns (Nucleosil C18 AB with octadecylsilica stationary phase, IAM PC C10/C3 with chemically bounded phosphatidylcholine, and Nucleosil 100-5 OH with chemically bounded propanodiole), and using isocratically acetonitrile: Britton-Robinson buffer as the mobile phase. Additionally, molecular modeling studies were performed with the use of HyperChem software and MM+ molecular mechanics with the semi-empirical AM1 method deriving 20 structural descriptors.