WHO Air Quality Guidelines, Global Update, 2005
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
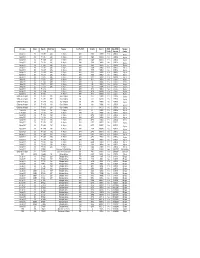
Final AFI RVSM Approvals 05 June 08
Mfr & Type Variant Reg. No. Build Year Operator Acft Op ICAO Serial No Mode S RVSM Date RVSM Operator Yes/No Approval Country Boeing 737 800 7T - VJK 2000 Air Algérie DAH 30203 0A0019 Yes 23/01/02 Algeria Boeing 737 800 7T - VJL 2000 Air Algérie DAH 30204 0A001A Yes 23/01/02 Algeria Boeing 737 800 7T - VJM 2000 Air Algérie DAH 30205 0A001B Yes 23/01/02 Algeria Boeing 737 800 7T - VJN 2000 Air Algérie DAH 30206 0A0020 Yes 23/01/02 Algeria Boeing 737 800 7T - VJQ 2002 Air Algérie DAH 30207 0A0021 Yes 23/01/02 Algeria Boeing 737 800 7T - VJP 2001 Air Algérie DAH 30208 0A0022 Yes 23/01/02 Algeria Boeing 737 600 7T - VJR 2002 Air Algérie DAH 30545 0A0025 Yes 01/06/02 Algeria Boeing 737 600 7T - VJS 2002 Air Algérie DAH 30210 0A0026 Yes 18/06/02 Algeria Boeing 737 600 7T - VJT 2002 Air Algérie DAH 30546 0A0027 Yes 18/06/02 Algeria Boeing 737 600 7T - VJU 2002 Air Algérie DAH 30211 0A0028 Yes 06/07/02 Algeria Airbus 330 202 7T - VJV 2005 Air Algérie DAH 0644 0A0044 Yes 31/01/05 Algeria Airbus 330 202 7T - VJW 2005 Air Algérie DAH 647 0A0045 Yes 05/03/05 Algeria Airbus 330 202 7T - VJY 2005 Air Algérie DAH 653 0A0047 Yes 20/03/05 Algeria Airbus 330 202 7T - VJX 2005 Air Algérie DAH 650 0A0046 Yes 20/03/05 Algeria Boeing 737 800 7T - VKA Air Algérie DAH 34164 0A0049 Yes 23/07/05 Algeria Boeing 737 800 7T - VKB Air Algérie DAH 34165 0A004A Yes 22/08/05 Algeria Boeing 737 800 7T - VKC Air Algérie DAH 34166 0A004B Yes 24/08/05 Algeria Gulfstream Aerospace SP 7T - VPC 2001 Gouv of Algeria IGA 1418 0A4009 Yes 27/07/05 Algeria Gulfstream Aerospace SP -

22 the East African Directorate of Civil Aviation
NOT FOR PUBLICATION INSTITUTE OF CURRENT WORLD AFFAIRS B- 67 Wsshlngton, D.C. ast Africa High Commission: November 2, 195/ (22) The ast African Directorate of Civil Aviation Mr. Walter S. Roers Institute of Current World Affairs 522 Fifth Avenue New York 6, New York Dear Mr. Rogers The considerable size of Best Afr, ica, with populated centers separated by wide tracts of rugged, poorly watered country through which road and rail routes are built with difficulty and then provide only slow service, gives air transport an important position in the economy of the area. Access to ast Africa from rope and elsewhere in the world is aso greatly enhanced by air transport, which need not follow the deviating contours of the continent. Businesses with b:'enches throughout @set Africa need fast assenger services to carry executives on supervisory visits; perishable commodities, important items for repair of key machlner, and )ivestock for breeding purposes provide further traffic; and a valuable tourist traffic is much dependent upon air transport. The direction and coordination of civil aviation, to help assure the quality and amplitude of aerodromes, aeraio directiona and communications methods, and aircraft safety standards, is an important responsibility which logically fsIs under a central authority. This central authority is the Directorate of Oivil Aviation, a department of the ast Africa High Commission. The Directorate, as an interterritorlal service already in existence, came under the administration of the High Oommisslon on its effective date of inception, January I, 98, an more specifically under the Commissioner for Transport, one of the four principal executive officers of the High Commission, on May I, 199. -

Strategies Deployed by Fly 540 Aviation Company To
STRATEGIES DEPLOYED BY FLY 540 AVIATION COMPANY TO SUSTAIN COMPETITIVE ADVANTAGE BY ANN WANJIRU GUANDARU A RESEARCH PROJECT SUBMITED IN PARTIAL FULLFILMENT OF THE REQUIREMENT FOR THE AWARD OF THE DEGREE OF MASTER OF BUSINESS ADMINISTRATION, SCHOOL OF BUSINESS, UNIVERSITY OF NAIROBI 2019 DECLARATION ii ACKNOWLEDGEMENT iii DEDICATION iv TABLE OF CONTENTS DECLARATION........................................................................................................................... ii ACKNOWLEDGEMENT ........................................................................................................... iii DEDICATION.............................................................................................................................. iv LIST OF ABBREVIATIONS/ACRONYMS ............................................................................. ix ABSTRACT ................................................................................................................................... x CHAPTER ONE: INTRODUCTION ......................................................................................... 1 1.1 Background of the Study .................................................................................. 1 1.1.1 Concept of Strategy ....................................................................................... 4 1.1.2 Competitive Advantage ................................................................................. 5 1.1.3 Aviation Industry in Kenya .......................................................................... -

Global Volatility Steadies the Climb
WORLD AIRLINER CENSUS Global volatility steadies the climb Cirium Fleet Forecast’s latest outlook sees heady growth settling down to trend levels, with economic slowdown, rising oil prices and production rate challenges as factors Narrowbodies including A321neo will dominate deliveries over 2019-2038 Airbus DAN THISDELL & CHRIS SEYMOUR LONDON commercial jets and turboprops across most spiking above $100/barrel in mid-2014, the sectors has come down from a run of heady Brent Crude benchmark declined rapidly to a nybody who has been watching growth years, slowdown in this context should January 2016 low in the mid-$30s; the subse- the news for the past year cannot be read as a return to longer-term averages. In quent upturn peaked in the $80s a year ago. have missed some recurring head- other words, in commercial aviation, slow- Following a long dip during the second half Alines. In no particular order: US- down is still a long way from downturn. of 2018, oil has this year recovered to the China trade war, potential US-Iran hot war, And, Cirium observes, “a slowdown in high-$60s prevailing in July. US-Mexico trade tension, US-Europe trade growth rates should not be a surprise”. Eco- tension, interest rates rising, Chinese growth nomic indicators are showing “consistent de- RECESSION WORRIES stumbling, Europe facing populist backlash, cline” in all major regions, and the World What comes next is anybody’s guess, but it is longest economic recovery in history, US- Trade Organization’s global trade outlook is at worth noting that the sharp drop in prices that Canada commerce friction, bond and equity its weakest since 2010. -

Evaristus M. Irandu University of Nairobi Nairobi, Kenya Dawna L
Journal of Air Transportation Vol. 11, No. 1 -2006 THE DEVELOPMENT OF JOMO KENYATTA INTERNATIONAL AIRPORT AS A REGIONAL AVIATION HUB Evaristus M. Irandu University of Nairobi Nairobi, Kenya Dawna L. Rhoades Embry-Riddle Aeronautical University Daytona Beach, Florida ABSTRACT Air transportation plays an important role in the social and economic development of the global system and the countries that seek to participate in it. As Africa seeks to take its place in the global economy, it is increasingly looking to aviation as the primary means of connecting its people and goods with the world. It has been suggested that Africa as a continent needs to move toward a system of hubs to optimize its scarce resources. Jomo Kenyatta International Airport in Nairobi, Kenya, is one of the airports in the eastern region of Africa that is seeking to fill this role. This paper discusses the prospects for success and the challenges that it will need to overcome, including projections through 2020 for the growth in passenger and cargo traffic. _____________________________________________________________________________ Evaristus M. Irandu received his Ph.D. in Transport Geography from the University of Nairobi. He is a Senior Lecturer and formerly, the Chairman of the Department of Geography and Environmental Studies in the University of Nairobi. He teaches Economic and Transport Geography, International Tourism and Tourism Management, at both undergraduate and graduate level. His research interests include: aviation planning, liberalization of air transport, non-motorized transport, urban transport, international tourism and ecotourism. His research work has appeared in several journals such as Anatolia: An International Journal of Tourism and Hospitality Research, Asia Pacific Journal of Tourism Research and Environment and Culture. -

Analysis of Global Airline Alliances As a Strategy for International Network Development by Antonio Tugores-García
Analysis of Global Airline Alliances as a Strategy for International Network Development by Antonio Tugores-García M.S., Civil Engineering, Enginyer de Camins, Canals i Ports Universitat Politècnica de Catalunya, 2008 Submitted to the MIT Engineering Systems Division and the Department of Aeronautics and Astronautics in Partial Fulfillment of the Requirements for the Degrees of Master of Science in Technology and Policy and Master of Science in Aeronautics and Astronautics at the Massachusetts Institute of Technology June 2012 © 2012 Massachusetts Institute of Technology. All rights reserved Signature of Author__________________________________________________________________________________ Antonio Tugores-García Department of Engineering Systems Division Department of Aeronautics and Astronautics May 14, 2012 Certified by___________________________________________________________________________________________ Peter P. Belobaba Principal Research Scientist, Department of Aeronautics and Astronautics Thesis Supervisor Accepted by__________________________________________________________________________________________ Joel P. Clark Professor of Material Systems and Engineering Systems Acting Director, Technology and Policy Program Accepted by___________________________________________________________________________________________ Eytan H. Modiano Professor of Aeronautics and Astronautics Chair, Graduate Program Committee 1 2 Analysis of Global Airline Alliances as a Strategy for International Network Development by Antonio Tugores-García -

Airlines Codes
Airlines codes Sorted by Airlines Sorted by Code Airline Code Airline Code Aces VX Deutsche Bahn AG 2A Action Airlines XQ Aerocondor Trans Aereos 2B Acvilla Air WZ Denim Air 2D ADA Air ZY Ireland Airways 2E Adria Airways JP Frontier Flying Service 2F Aea International Pte 7X Debonair Airways 2G AER Lingus Limited EI European Airlines 2H Aero Asia International E4 Air Burkina 2J Aero California JR Kitty Hawk Airlines Inc 2K Aero Continente N6 Karlog Air 2L Aero Costa Rica Acori ML Moldavian Airlines 2M Aero Lineas Sosa P4 Haiti Aviation 2N Aero Lloyd Flugreisen YP Air Philippines Corp 2P Aero Service 5R Millenium Air Corp 2Q Aero Services Executive W4 Island Express 2S Aero Zambia Z9 Canada Three Thousand 2T Aerocaribe QA Western Pacific Air 2U Aerocondor Trans Aereos 2B Amtrak 2V Aeroejecutivo SA de CV SX Pacific Midland Airlines 2W Aeroflot Russian SU Helenair Corporation Ltd 2Y Aeroleasing SA FP Changan Airlines 2Z Aeroline Gmbh 7E Mafira Air 3A Aerolineas Argentinas AR Avior 3B Aerolineas Dominicanas YU Corporate Express Airline 3C Aerolineas Internacional N2 Palair Macedonian Air 3D Aerolineas Paraguayas A8 Northwestern Air Lease 3E Aerolineas Santo Domingo EX Air Inuit Ltd 3H Aeromar Airlines VW Air Alliance 3J Aeromexico AM Tatonduk Flying Service 3K Aeromexpress QO Gulfstream International 3M Aeronautica de Cancun RE Air Urga 3N Aeroperlas WL Georgian Airlines 3P Aeroperu PL China Yunnan Airlines 3Q Aeropostal Alas VH Avia Air Nv 3R Aerorepublica P5 Shuswap Air 3S Aerosanta Airlines UJ Turan Air Airline Company 3T Aeroservicios -

Airline Route Networks: a Complex Network Approach C February 2014 Als Meus Pares
ADVERTIMENT . La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del servei TDX ( www.tesisenxarxa.net ) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei TDX. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a TDX (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA . La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del servicio TDR ( www.tesisenred.net ) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio TDR. No se autoriza la presentación de su contenido en una ventana o marco ajeno a TDR (framing). Esta reserva de derechos afecta tanto al resumen de presentación de la tesis como a sus contenidos. En la utilización o cita de partes de la tesis es obligado indicar el nombre de la persona autora. -

Strengthening Cooperation with Regional Air Pollution Networks and Initiatives Outside the Convention Explanatory Note to the Do
Executive Body, twenty‐ninth session, Geneva, 12 – 16 December 2011 Informal document No.12 Strengthening Cooperation with Regional Air Pollution Networks and Initiatives outside the Convention Submitted by the secretariat of the Global Atmospheric Pollution Forum Explanatory note to the document The Global Atmospheric Pollution Forum brings together regional intergovernmental networks and other governmental and non-governmental bodies concerned with cooperation on air pollution at the regional, hemispheric and global scales. Its aim is to promote cooperative action among them, and more generally to enhance capacity to address air pollution issues at regional, hemispheric and global scales. The Forum is currently undertaking a wide-ranging review of inter-regional cooperation on air pollution which it expects to publish in the earlier part of 2012. This paper, which summarizes assessments prepared so far as part of that review, and some of their implications and conclusions, provides background information for the Executive Body and other bodies of the Convention. It aims to help consideration of how to give further effect to the commitment to “outreach” and to closer cooperation with intergovernmental air pollution networks in other regions. 1 Executive Body, twenty‐ninth session, Geneva, 12 – 16 December 2011 Informal document No.12 Strengthening Cooperation with Regional Air Pollution Networks and Initiatives outside the Convention Submitted by the secretariat of the Global Atmospheric Pollution Forum Summary This document aims to aid consideration of future broader cooperation with regional efforts outside the Convention as required by the Convention’s long-term strategy. It summarizes background information on existing regional networks and agreements, including information on membership, legal status, structures, current activities and existing links with the Convention. -

Draft Sadc Regional Policy Framework on Air Pollution
DRAFT SADC REGIONAL POLICY FRAMEWORK ON AIR POLLUTION Lusaka Agreement (2008) - Southern African Development Community (SADC) Regional Policy Framework on Air Pollution Participants from the 14 Southern African Development Community (SADC) countries, representing governments, industry, non-governmental organizations (NGOs), civil society, international organizations and academia, attended the Southern Africa Sub-Regional Policy Dialogue on Air Pollution in Lusaka, Zambia, during 5-6 March 2008 to develop the following Regional Policy Framework on Air Pollution in SADC that was subsequently adopted by SADC Ministers at the Ministerial Session held on 7 March 2008. Considering: The significant and increasing impacts that air pollutants can have on health, crops, natural ecosystems, man-made materials and visibility, and their socio- economic consequences for SADC countries. The most relevant indoor and outdoor air pollutants, that cause significant damage locally, nationally and in a transboundary context, in the SADC region are particulate matter (PM – the most health damaging components characterised as PM2.5); sulphur dioxide (SO2); nitrogen dioxide (NO2); ammonia (NH3); volatile organic compounds (VOCs); persistent organic pollutants (POPs), such as dioxins and furans; heavy metals, such as arsenic (As), cadmium (Cd), mercury (Hg) and lead (Pb); and fluorides. The strong linkages between emissions of air pollutants and greenhouse gases and the co-benefits of reducing air pollution in all sectors for greenhouse gas emission reductions, reducing congestion etc. The linkages to the production of secondary pollutants, such as ozone, that can have significant impacts on health and crop yield and quality. Recognizing that: Air pollution takes a high toll on the health, environment and economies of African countries, and that the transport sector in particular is responsible for rapidly increasing vehicle emissions from the use of high sulphur fuels and older motor vehicles, especially of particulate matter – a major global health concern. -

Improving Flight Connections in Ecowas Region
IMPROVING FLIGHT CONNECTIONS IN ECOWAS REGION Presented by Dr. Paul-Antoine Marie GANEMTORE Head of Air Transport Unit Infrastructure Department ECOWAS COMMISSION 1 Presentation Outline CHALLENGES OF AIR TRANSPORT COMMUNITY AIR TRANSPORT LEGAL FRAMEWORK WAY FORWARD CHALLENGES OF AIR TRANSPORT 3 CONTEXT CREATION 28 May, 1975 in Lagos, Nigeria, the Economic Community of West African States (ECOWAS) OBJECTIVE Co-operation and integration to support growth in regional trade and free movement, leading to establishment of an Economic Union in West Africa Article 32 –f: “Encourage co-operation in flight-scheduling, leasing of aircraft and granting and joint use of fifth freedom rights to airlines of the region” 15 MEMBER STATES Benin, Burkina Faso, Cape Verde, Côte d’Ivoire, Gambia, Ghana, Guinea, Guinea Bissau, Liberia, Mali, Niger, Nigeria, Senegal, Sierra Leone & Togo GENERAL DATA 328 Million inhabitants in 5,1 Million Km2 surface area 534 Billion USD GDP Mineral Resources (Petrol, Gas, Gold, Uranium, Phosphate,...) & Agriculture Resources(Cocoa, Coffee, Sugar, Cotton, Rubber, Wood...) ROLE OF ECOWAS COMMISSION Coordination and harmonization of policy, programmes, actions, projets, as entrhrusted by the member States….. 4 2013 ECOWAS AIR TRANSPORT MARKET YEAR 2013 PAX CARGO AIRCRAFT POPULATION SURFACE GDP m Ton mvts m Km2 Billions BENIN 476,704 7,616 12,309 10,300,000 112,760 8.30 BURKINA FASO 523,355 7,011 9,936 17,800,000 273,600 12.2 CAPE VERDE 1,957,747 3,061.5 28,702 0,530,000 4,030 1.9 COTE D’IVOIRE 1,152,887 17,548 18,195 -

Opening African Skies: the Case of Airline Industry Liberalization in East Africa Author(S): Evaristus M
Transportation Research Forum Opening African Skies: The Case of Airline Industry Liberalization in East Africa Author(s): Evaristus M. Irandu Source: Journal of the Transportation Research Forum, Vol. 47, No. 1 (Spring 2008), pp. 73-88 Published by: Transportation Research Forum Stable URL: http://www.trforum.org/journal The Transportation Research Forum, founded in 1958, is an independent, nonprofit organization of transportation professionals who conduct, use, and benefit from research. Its purpose is to provide an impartial meeting ground for carriers, shippers, government officials, consultants, university researchers, suppliers, and others seeking exchange of information and ideas related to both passenger and freight transportation. More information on the Transportation Research Forum can be found on the Web at www.trforum.org. OPENING AFRICAN SKIES: THE CASE OF AIRLINE INDUSTRY LIBERALIZATION IN EAST AFRICA by Evaristus M. Irandu The three member states of the East African Community (EAC) have made great efforts in modernizing their air transport industry in order to meet the increased demand for international tourism and horticultural export trade. The policy adopted by the sub-region, at independence, to regulate the airline industry was the bilateralism approach with emphasis on reciprocity. Such a policy became a bottleneck in the development of the air transport industry in the sub-region. It became apparent that the EAC member states needed efficient air services and not airlines that served mainly as status symbols. However, the last decade witnessed the liberalization of the aviation sector in the sub-region. This liberalization process is part of a proactive policy to encourage global investment in the sub-region.