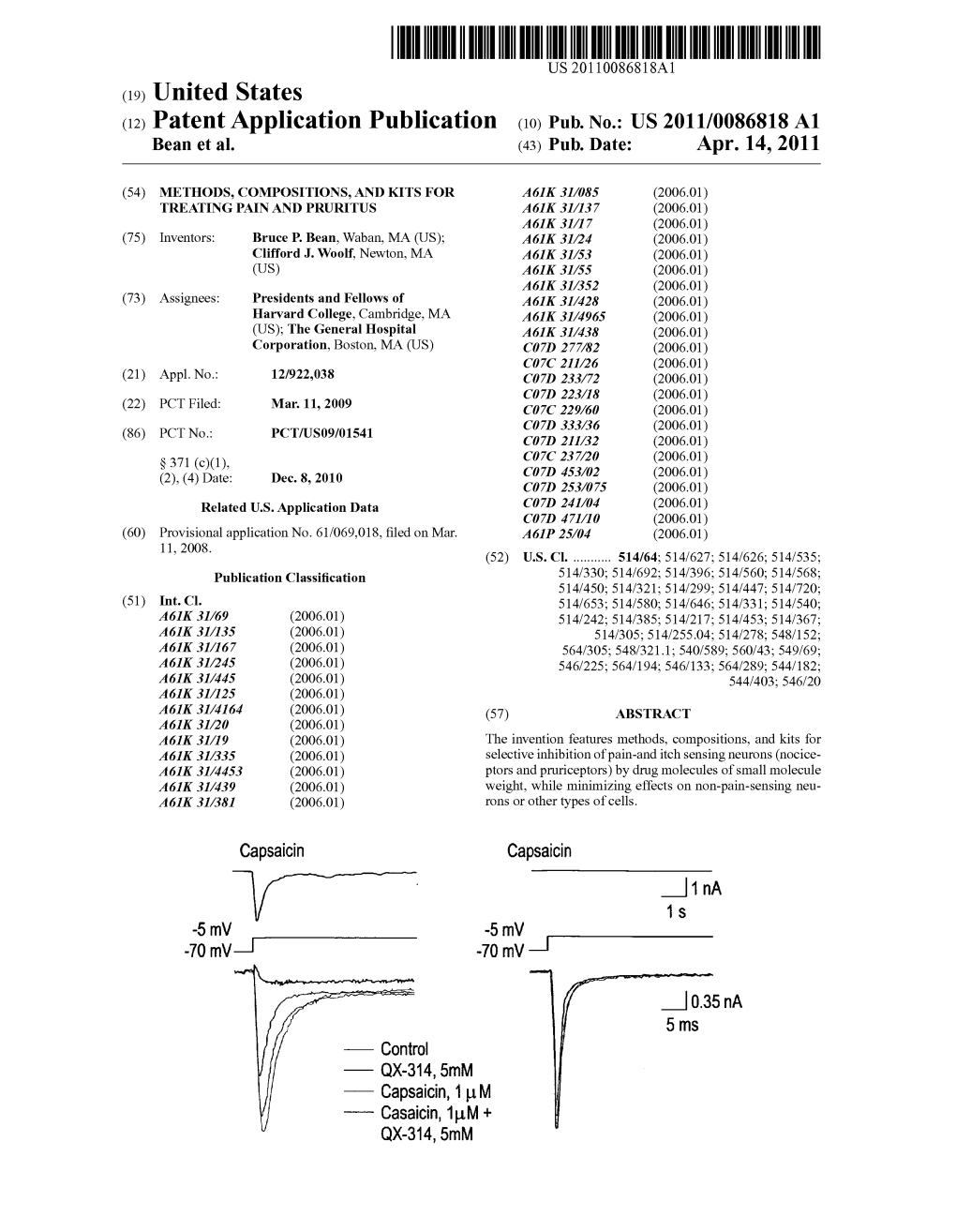
(12) Patent Application Publication (10) Pub. No.: US 2011/0086818 A1 Bean Et Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Europæisk Patentskrift
(19) DANMARK (1°) DK/EP 2968225 T3 (12) Oversættelse af europæisk patentskrift Patent- og Varemærkestyrelsen (51) lnt.CI.: A 61 K 31/137 (2006.01) A 61 K 9/00 (2006.01) A 61 K 31/245 (2006.01) A 61 K 31/445 (2006.01) A 61 K 31/519 (2006.01) A 61 K 31/52 (2006.01) A 61 P 43/00 (2006.01) (45) Oversættelsen bekendtgjort den: 2019-05-27 (80) Dato for Den Europæiske Patentmyndigheds bekendtgørelse om meddelelse af patentet: 2019-02-20 (86) Europæisk ansøgning nr.: 14719486.4 (86) Europæisk indleveringsdag: 2014-03-17 (87) Den europæiske ansøgnings publiceringsdag: 2016-01-20 (86) International ansøgning nr.: US2014030372 (87) Internationalt publikationsnr.: WO2014145580 (30) Prioritet: 2013-03-15 US 201361789054 P (84) Designerede stater: AL AT BE BG CH CY CZ DE DK EE ES Fl FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (73) Patenthaver: The Children's Medical Center Corporation, 55 Shattuck Street, Boston, Massachusetts 02115, USA (72) Opfinder: BERDE, Charles, 14 Doran Road, Bookline, MA 02146, USA KOHANE, Daniel S., 119 Willard Street, Newton, MA 02461, USA (74) Fuldmægtig i Danmark: AWA Denmark A/S, Strandgade 56,1401 København K, Danmark (54) Benævnelse: NEOSAXITOXINKOMBINATIONSFORMULERINGER TIL FORLÆNGET LOKALANÆSTESI (56) Fremdragne publikationer: WO-A2-98/51290 ALBERTO J. RODRIGUEZ-NAVARRO ET AL: "Potentiation of Local Anesthetic Activity of Neosaxitoxin with Bupivacaine or Epinephrine: Development of a Long-Acting Pain Blocker", NEUROTOXICITY RESEARCH, vol. 16, no. 4, 28 July 2009 (2009-07-28), pages 408-415, XP055122925, ISSN: 1029-8428, DOI: 10.1007/s12640-009- 9092-3 cited in the application Anonymous: "NCT01786655 on 2013_02_11: ClinicalTrials.gov Archive",, 11 February 2013 (2013-02-11), XP055122935, Retrieved from the Internet: URL:http://clinicaltrials.gov/archive/NCTO 1786655/2013_02_11 [retrieved on 2014-06-12] CHARLES B. -

TRP Mediation
molecules Review Remedia Sternutatoria over the Centuries: TRP Mediation Lujain Aloum 1 , Eman Alefishat 1,2,3 , Janah Shaya 4 and Georg A. Petroianu 1,* 1 Department of Pharmacology, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates; [email protected] (L.A.); Eman.alefi[email protected] (E.A.) 2 Center for Biotechnology, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates 3 Department of Biopharmaceutics and Clinical Pharmacy, Faculty of Pharmacy, The University of Jordan, Amman 11941, Jordan 4 Pre-Medicine Bridge Program, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates; [email protected] * Correspondence: [email protected]; Tel.: +971-50-413-4525 Abstract: Sneezing (sternutatio) is a poorly understood polysynaptic physiologic reflex phenomenon. Sneezing has exerted a strange fascination on humans throughout history, and induced sneezing was widely used by physicians for therapeutic purposes, on the assumption that sneezing eliminates noxious factors from the body, mainly from the head. The present contribution examines the various mixtures used for inducing sneezes (remedia sternutatoria) over the centuries. The majority of the constituents of the sneeze-inducing remedies are modulators of transient receptor potential (TRP) channels. The TRP channel superfamily consists of large heterogeneous groups of channels that play numerous physiological roles such as thermosensation, chemosensation, osmosensation and mechanosensation. Sneezing is associated with the activation of the wasabi receptor, (TRPA1), typical ligand is allyl isothiocyanate and the hot chili pepper receptor, (TRPV1), typical agonist is capsaicin, in the vagal sensory nerve terminals, activated by noxious stimulants. -

Pharmacokinetics of Daikenchuto, a Traditional Japanese Medicine (Kampo) After Single Oral Administration to Healthy Japanese Volunteers
DMD Fast Forward. Published on July 1, 2011 as DOI: 10.1124/dmd.111.040097 DMDThis Fast article Forward. has not been Published copyedited and on formatted. July 1, The2011 final as version doi:10.1124/dmd.111.040097 may differ from this version. DMD #040097 Pharmacokinetics of daikenchuto, a traditional Japanese medicine (Kampo) after single oral administration to healthy Japanese volunteers Masaya Munekage, Hiroyuki Kitagawa, Kengo Ichikawa, Junko Watanabe, Katsuyuki Aoki, Toru Kono, Kazuhiro Hanazaki Department of Surgery, Kochi Medical School, Nankoku, Kochi, Japan (M.M., H.K., K.I., K.H); Downloaded from Tsumura Laboratories, TSUMURA & CO., Ami, Ibaraki, Japan (J.W.); Pharmaceutical & Quality Research Department, TSUMURA & CO., Ami, Ibaraki , Japan (K.A.); Division of dmd.aspetjournals.org Gastroenterologic and General Surgery, Department of Surgery, Asahikawa Medical University, Hokkaido, Japan (T.K.). at ASPET Journals on September 26, 2021 1 Copyright 2011 by the American Society for Pharmacology and Experimental Therapeutics. DMD Fast Forward. Published on July 1, 2011 as DOI: 10.1124/dmd.111.040097 This article has not been copyedited and formatted. The final version may differ from this version. DMD #040097 Running title: Pharmacokinetics study of daikenchuto Address correspondence to: Kazuhiro Hanazaki, M.D., Ph.D. Department of Surgery, Kochi Medical School, Oko-cho kohasu, Nankoku-shi, Kochi 783-8505, Japan. E-mail: [email protected] , Phone: 81-88-880-2370, Fax: 81-88-880-2371 Number of text pages: 17 Downloaded from Number of Tables: 1 Number of Figures: 2 dmd.aspetjournals.org Number of References: 17 Number of Words: Abstract: 199 at ASPET Journals on September 26, 2021 Introduction: 377 Results and Discussion: 855 ABBREVIATIONS: TJ-100, daikenchuto; HAS, hydroxy-α-sanshool; HBS, hydroxy-β-sanshool; 6S, [6]-shogaol; 10S, [10]-shogaol; GRB1, ginsenoside Rb1; GRG1, ginsenoside Rg1; HPLC, high-performance liquid chromatography; LC, liquid chromatography; MS, mass spectrometry; MS/MS, tandem mass spectrometry 2 DMD Fast Forward. -

Less Than Lethal Weapons
PUBLIC ORDER MANAGEMENT Less Than Lethal Weapons UN Peacekeeping PDT Standards for Formed Police Units 1st edition 2015 Public Order Management 1 Less Than Lethal Weapons Background Before the inception of UN Peacekeeping mission, the Department of Peacekeeping Operations requests TCC/PCC to contribute with their forces to the strength of the mission. The UN Police component is composed by Individual Police Officers (IPO) and Formed Police Units (FPU). The deployment of FPU is subject to a Memorandum of Understanding between the UN and the contributing country and the compliance with the force requirements of the mission. The force requirement lists the equipment and the weapons that the FPU has to deploy with. Despite the fact ‘Guidelines on the Use of Force by Law Enforcement Agencies’ recommends the development and the deployment of less than lethal weapons and ammunitions, FPUs usually do not possess this type of equipment. Until the development of less-lethal weapons, police officers around the world had few if any less-lethal options for riot control. Common tactics used by police that were intended to be non-lethal or less than lethal included a slowly advancing wall of men with batons. Considering the tasks the FPUs are demanded to carry out, those weapons should be mandatory as part of their equipment. The more equipped with these weapons FPUs are, the more they will be able to efficiently respond to the different type of threats and situation. Non-lethal weapons, also called less-lethal weapons, less-than-lethal weapons, non- deadly weapons, compliance weapons, or pain-inducing weapons are weapons intended to be used in the scale of Use of Force before using any lethal weapon. -

Classification of Medicinal Drugs and Driving: Co-Ordination and Synthesis Report
Project No. TREN-05-FP6TR-S07.61320-518404-DRUID DRUID Driving under the Influence of Drugs, Alcohol and Medicines Integrated Project 1.6. Sustainable Development, Global Change and Ecosystem 1.6.2: Sustainable Surface Transport 6th Framework Programme Deliverable 4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Due date of deliverable: 21.07.2011 Actual submission date: 21.07.2011 Revision date: 21.07.2011 Start date of project: 15.10.2006 Duration: 48 months Organisation name of lead contractor for this deliverable: UVA Revision 0.0 Project co-funded by the European Commission within the Sixth Framework Programme (2002-2006) Dissemination Level PU Public PP Restricted to other programme participants (including the Commission x Services) RE Restricted to a group specified by the consortium (including the Commission Services) CO Confidential, only for members of the consortium (including the Commission Services) DRUID 6th Framework Programme Deliverable D.4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Page 1 of 243 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Authors Trinidad Gómez-Talegón, Inmaculada Fierro, M. Carmen Del Río, F. Javier Álvarez (UVa, University of Valladolid, Spain) Partners - Silvia Ravera, Susana Monteiro, Han de Gier (RUGPha, University of Groningen, the Netherlands) - Gertrude Van der Linden, Sara-Ann Legrand, Kristof Pil, Alain Verstraete (UGent, Ghent University, Belgium) - Michel Mallaret, Charles Mercier-Guyon, Isabelle Mercier-Guyon (UGren, University of Grenoble, Centre Regional de Pharmacovigilance, France) - Katerina Touliou (CERT-HIT, Centre for Research and Technology Hellas, Greece) - Michael Hei βing (BASt, Bundesanstalt für Straßenwesen, Germany). -

Medicines Used Across Wirral Health Economy
Medicines used across Wirral Health Economy Below is a list of medicines that are approved for use on Wirral. Some medicines may only be prescribed in hospital, others may be prescribed by your GP. The list is updated every time a new medicine is approved for use in Wirral. Some medicines have been recommended by the National Institute for Health and Care Excellence (NICE). The middle column of the table below indicates recommendations made by NICE — eg, TA234 means that the medicine was reviewed in NICE technology appraisal number 234. Some of the medicines we use are recommended by specialists working from the Walton Centre NHS Foundation Trust or from the Cheshire and Wirral Partnership NHS Foundation Trust. These medicines are included in our formulary — however; the prescriber retains responsibility for the appropriate use of these medicines. These formularies are obtainable here: http://www.panmerseyapc.nhs.uk/formulary/documents/04- 0800_antiepileptic_drugs.pdf http://www.panmerseyapc.nhs.uk/formulary/documents/04-09- 00_parkinsonism.pdf http://www.cwp.nhs.uk/pages/629-pharmacy-and-medicines-information Subject to NICE Drug Name recommendation Abatacept TA195, TA280 Acamprosate Acarbose Accu-Chek Inform II® AccuSol® 35 Potassium 4mmol/L Acetazolamide Acetylcysteine Acencoumarol Acetic acid 5% solution Acetone Acitretin Aciclovir Wirral Medicines Formulary Author: Medicines Management Team Approved by: Wirral Drug and Therapeutics Panel Last updated: June 2015 Version 10 Aclinidium Actilite® Activated Charcoal Activon® - see -

Proxymetacaine Hydrochloride Ropivacaine Hydrochloride
1870 Local Anaesthetics Preparations pore: Alcaine; Switz.: Alcaine; Turk.: Alcaine; Opticaine; USA: Ak-Taine; Alcaine; Ocu-Caine; Ophthetic; Parcaine; Venez.: Alcaine; Oftaine†; Poen- (details are given in Part 3) caina. Proprietary Preparations CH3 Multi-ingredient: Spain: Detraine. O Multi-ingredient: Canad.: Fluoracaine†; USA: Fluoracaine; Fluorocaine. N CH3 O Quinisocaine Hydrochloride (BANM, rINNM) H3C Propipocaine (rINN) O Chinisocainum Hydrochloride; Dimethisoquin Hydrochloride Propipocaína; Propipocaïne; Propipocainum; Propoxypipero- (USAN); Dimethisoquinium Chloride; Hidrocloruro de quinisocaí- NH2 caine. 3-Piperidino-4′-propoxypropiophenone. na; Quinisocaïne, Chlorhydrate de; Quinisocaini Hydrochlori- (proxymetacaine) dum. 2-(3-Butyl-1-isoquinolyloxy)-NN-dimethylethylamine hy- Пропипокаин drochloride. C17H25NO2 = 275.4. Хинизокаина Гидрохлорид CAS — 3670-68-6. NOTE. PROX is a code approved by the BP 2008 for use on single unit doses of eye drops containing proxymetacaine hydrochlo- C17H24N2O,HCl = 308.8. ride where the individual container may be too small to bear all CAS — 86-80-6 (quinisocaine); 2773-92-4 (quinisocaine the appropriate labelling information. PROXFLN is a similar hydrochloride). O code approved for eye drops containing proxymetacaine hydro- ATC — D04AB05. chloride and fluorescein sodium. ATC Vet — QD04AB05. N Pharmacopoeias. In Br. and US. BP 2008 (Proxymetacaine Hydrochloride). A white or almost H3C CH3 white, odourless or almost odourless, crystalline powder. Soluble O in water and in chloroform; very soluble in dehydrated alcohol; N practically insoluble in ether. A 1% solution in water has a pH of O CH Profile 5.7 to 6.4. Protect from light. 3 Propipocaine is a local anaesthetic (p.1850) that has been used USP 31 (Proparacaine Hydrochloride). A white to off-white, or for surface anaesthesia. -
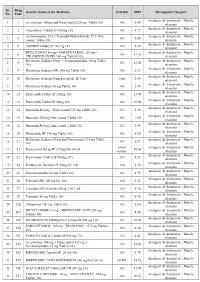
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

TRP Channel Transient Receptor Potential Channels
TRP Channel Transient receptor potential channels TRP Channel (Transient receptor potential channel) is a group of ion channels located mostly on the plasma membrane of numerous human and animal cell types. There are about 28 TRP channels that share some structural similarity to each other. These are grouped into two broad groups: Group 1 includes TRPC ("C" for canonical), TRPV ("V" for vanilloid), TRPM ("M" for melastatin), TRPN, and TRPA. In group 2, there are TRPP ("P" for polycystic) and TRPML ("ML" for mucolipin). Many of these channels mediate a variety of sensations like the sensations of pain, hotness, warmth or coldness, different kinds of tastes, pressure, and vision. TRP channels are relatively non-selectively permeable to cations, including sodium, calcium and magnesium. TRP channels are initially discovered in trp-mutant strain of the fruit fly Drosophila. Later, TRP channels are found in vertebrates where they are ubiquitously expressed in many cell types and tissues. TRP channels are important for human health as mutations in at least four TRP channels underlie disease. www.MedChemExpress.com 1 TRP Channel Inhibitors, Antagonists, Agonists, Activators & Modulators (-)-Menthol (E)-Cardamonin Cat. No.: HY-75161 ((E)-Cardamomin; (E)-Alpinetin chalcone) Cat. No.: HY-N1378 (-)-Menthol is a key component of peppermint oil (E)-Cardamonin ((E)-Cardamomin) is a novel that binds and activates transient receptor antagonist of hTRPA1 cation channel with an IC50 potential melastatin 8 (TRPM8), a of 454 nM. Ca2+-permeable nonselective cation channel, to 2+ increase [Ca ]i. Antitumor activity. Purity: >98.0% Purity: 99.81% Clinical Data: Launched Clinical Data: No Development Reported Size: 10 mM × 1 mL, 500 mg, 1 g Size: 10 mM × 1 mL, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg (Z)-Capsaicin 1,4-Cineole (Zucapsaicin; Civamide; cis-Capsaicin) Cat. -

Ep 1931310 B1
(19) & (11) EP 1 931 310 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 9/12 (2006.01) A61K 9/06 (2006.01) 06.06.2012 Bulletin 2012/23 A61K 9/70 (2006.01) A61K 47/32 (2006.01) A61K 47/24 (2006.01) A61K 47/10 (2006.01) (21) Application number: 06779420.6 (86) International application number: (22) Date of filing: 14.09.2006 PCT/GB2006/003408 (87) International publication number: WO 2007/031753 (22.03.2007 Gazette 2007/12) (54) Monophasic film-forming composition for topical administration Monophasische filmbildende Zusammensetzung zum topischen Auftrag Composition monophase formant un film pour l’administration à voie topique (84) Designated Contracting States: • JONES, Stuart, Allen AT BE BG CH CY CZ DE DK EE ES FI FR GB GR London SE3 0XA (GB) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR (74) Representative: Lord, Hilton David Marks & Clerk LLP (30) Priority: 14.09.2005 GB 0518769 90 Long Acre London (43) Date of publication of application: WC2E 9RA (GB) 18.06.2008 Bulletin 2008/25 (56) References cited: (73) Proprietor: Medpharm Limited WO-A2-01/43722 US-A- 4 752 466 Charlbury, US-A- 4 863 721 US-A1- 2004 184 994 Oxfordshire OX7 3RR (GB) US-A1- 2004 213 744 (72) Inventors: • BROWN, Marc, Barry Hertfordshire WD19 4QQ (GB) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

8–16–01 Vol. 66 No. 159 Thursday Aug. 16, 2001 Pages 42929
8–16–01 Thursday Vol. 66 No. 159 Aug. 16, 2001 Pages 42929–43064 VerDate 11-MAY-2000 19:13 Aug 15, 2001 Jkt 194001 PO 00000 Frm 00001 Fmt 4710 Sfmt 4710 E:\FR\FM\16AUWS.LOC pfrm11 PsN: 16AUWS 1 II Federal Register / Vol. 66, No. 159 / Thursday, August 16, 2001 The FEDERAL REGISTER is published daily, Monday through SUBSCRIPTIONS AND COPIES Friday, except official holidays, by the Office of the Federal Register, National Archives and Records Administration, PUBLIC Washington, DC 20408, under the Federal Register Act (44 U.S.C. Subscriptions: Ch. 15) and the regulations of the Administrative Committee of Paper or fiche 202–512–1800 the Federal Register (1 CFR Ch. I). The Superintendent of Assistance with public subscriptions 512–1806 Documents, U.S. Government Printing Office, Washington, DC 20402 is the exclusive distributor of the official edition. General online information 202–512–1530; 1–888–293–6498 Single copies/back copies: The Federal Register provides a uniform system for making available to the public regulations and legal notices issued by Paper or fiche 512–1800 Federal agencies. These include Presidential proclamations and Assistance with public single copies 512–1803 Executive Orders, Federal agency documents having general FEDERAL AGENCIES applicability and legal effect, documents required to be published Subscriptions: by act of Congress, and other Federal agency documents of public interest. Paper or fiche 523–5243 Assistance with Federal agency subscriptions 523–5243 Documents are on file for public inspection in the Office of the Federal Register the day before they are published, unless the issuing agency requests earlier filing. -

WO 2016/140629 Al 9 September 2016 (09.09.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/140629 Al 9 September 2016 (09.09.2016) P O P C T (51) International Patent Classification: (74) Agent: BULUT, Pinar; FarmaPatent Limited Sirketi, A61K 36/28 (2006.01) A61P 17/02 (2006.01) GMK Bulvari No:42/5, Maltepe, 06440 Ankara (TR). A61K 36/38 (2006.01) A61P 23/00 (2006.01) (81) Designated States (unless otherwise indicated, for every A61K 31/167 (2006.01) kind of national protection available): AE, AG, AL, AM, (21) International Application Number: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, PCT/TR20 15/000088 BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 5 March 2015 (05.03.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (25) Filing Language: English MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (71) Applicant: PHARMACTFVE ILAC SANAYI VE Tl- TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. CARET A. S. [TR/TR]; Mahmutbey Mah. Dilmenler Cad., (84) Designated States (unless otherwise indicated, for every Aslanoba Plaza No: 19 K:3, Bagcilar/istanbul (TR).