Documento Completo Descargar Archivo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
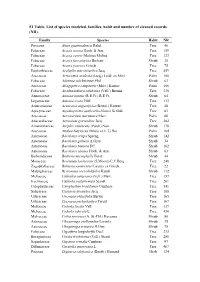
S1 Table. List of Species Modeled, Families, Habit and Number of Cleaned Records (NR)
S1 Table. List of species modeled, families, habit and number of cleaned records (NR). Family Species Habit NR Pinaceae Abies guatemalensis Rehd. Tree 46 Fabaceae Acacia aroma Hook. & Arn. Tree 159 Fabaceae Acacia caven (Molina) Molina Tree 123 Fabaceae Acacia furcatispina Burkart Shrub 35 Fabaceae Acacia praecox Griseb. Tree 75 Euphorbiaceae Acalypha macrostachya Jacq. Tree 459 Arecaceae Acrocomia aculeata (Jacq.) Lodd. ex Mart. Palm 166 Fabaceae Adesmia volckmannii Phil. Shrub 63 Arecaceae Allagoptera campestris (Mart.) Kuntze Palm 106 Fabaceae Anadenanthera colubrina (Vell.) Brenan Tree 330 Annonaceae Annona nutans (R.E.Fr.) R.E.Fr. Shrub 64 Loganiaceae Antonia ovata Pohl Tree 113 Araucariaceae Araucaria angustifolia (Bertol.) Kuntze Tree 48 Apocynaceae Aspidosperma quebracho-blanco Schltdl. Tree 83 Arecaceae Astrocaryum murumuru Mart. Palm 66 Anacardiaceae Astronium graveolens Jacq. Tree 282 Amaranthaceae Atriplex canescens (Pursh) Nutt. Shrub 178 Arecaceae Attalea butyracea (Mutis ex L.f.) We Palm 104 Asteraceae Baccharis crispa Spreng. Shrub 142 Asteraceae Baccharis gilliesii A.Gray Shrub 34 Asteraceae Baccharis trimera DC. Shrub 162 Asteraceae Baccharis ulicina Hook. & Arn. Shrub 63 Berberidaceae Berberis microphylla Forst. Shrub 44 Moraceae Brosimum lactescens (S.Moore) C.C.Berg Tree 248 Zygophyllaceae Bulnesia sarmientoi Lorentz ex Griseb. Tree 22 Malpighiaceae Byrsonima coccolobifolia Kunth Shrub 112 Meliaceae Cabralea canjerana (Vell.) Mart. Tree 157 Icacinaceae Calatola costaricensis Standl. Tree 201 Calophyllaceae Calophyllum brasiliense Cambess. Tree 541 Salicaceae Casearia decandra Jacq. Tree 188 Urticaceae Cecropia obtusifolia Bertol. Tree 165 Urticaceae Cecropia pachystachya Trécul Tree 167 Meliaceae Cedrela fissilis Vell. Tree 137 Meliaceae Cedrela odorata L. Tree 436 Malvaceae Ceiba speciosa (A. St.-Hil.) Ravenna Shrub 80 Asteraceae Chuquiraga avellanedae Lorentz Shrub 35 Asteraceae Chuquiraga erinacea D.Don Shrub 75 Fabaceae Copaifera langsdorffii Desf. -

Supplementary Materialsupplementary Material
10.1071/BT13149_AC © CSIRO 2013 Australian Journal of Botany 2013, 61(6), 436–445 SUPPLEMENTARY MATERIAL Comparative dating of Acacia: combining fossils and multiple phylogenies to infer ages of clades with poor fossil records Joseph T. MillerA,E, Daniel J. MurphyB, Simon Y. W. HoC, David J. CantrillB and David SeiglerD ACentre for Australian National Biodiversity Research, CSIRO Plant Industry, GPO Box 1600 Canberra, ACT 2601, Australia. BRoyal Botanic Gardens Melbourne, Birdwood Avenue, South Yarra, Vic. 3141, Australia. CSchool of Biological Sciences, Edgeworth David Building, University of Sydney, Sydney, NSW 2006, Australia. DDepartment of Plant Biology, University of Illinois, Urbana, IL 61801, USA. ECorresponding author. Email: [email protected] Table S1 Materials used in the study Taxon Dataset Genbank Acacia abbreviata Maslin 2 3 JF420287 JF420065 JF420395 KC421289 KC796176 JF420499 Acacia adoxa Pedley 2 3 JF420044 AF523076 AF195716 AF195684; AF195703 Acacia ampliceps Maslin 1 KC421930 EU439994 EU811845 Acacia anceps DC. 2 3 JF420244 JF420350 JF419919 JF420130 JF420456 Acacia aneura F.Muell. ex Benth 2 3 JF420259 JF420036 JF420366 JF419935 JF420146 KF048140 Acacia aneura F.Muell. ex Benth. 1 2 3 JF420293 JF420402 KC421323 JQ248740 JF420505 Acacia baeuerlenii Maiden & R.T.Baker 2 3 JF420229 JQ248866 JF420336 JF419909 JF420115 JF420448 Acacia beckleri Tindale 2 3 JF420260 JF420037 JF420367 JF419936 JF420147 JF420473 Acacia cochlearis (Labill.) H.L.Wendl. 2 3 KC283897 KC200719 JQ943314 AF523156 KC284140 KC957934 Acacia cognata Domin 2 3 JF420246 JF420022 JF420352 JF419921 JF420132 JF420458 Acacia cultriformis A.Cunn. ex G.Don 2 3 JF420278 JF420056 JF420387 KC421263 KC796172 JF420494 Acacia cupularis Domin 2 3 JF420247 JF420023 JF420353 JF419922 JF420133 JF420459 Acacia dealbata Link 2 3 JF420269 JF420378 KC421251 KC955787 JF420485 Acacia dealbata Link 2 3 KC283375 KC200761 JQ942686 KC421315 KC284195 Acacia deanei (R.T.Baker) M.B.Welch, Coombs 2 3 JF420294 JF420403 KC421329 KC955795 & McGlynn JF420506 Acacia dempsteri F.Muell. -

The Genus Anadenanthera in Amerindian Cultures
THE GENUS ANADENANTHERA IN AMERINDIAN CULTURES BY SIRI VON REIS ALTSCHUL, PH .D. RESEARCH FELLOW BOTANICAL MUSEUM HARVARD UNIVERSITY CAMBRIDGE, MASSACHUSETTS 1972 This monograph is dedicated in affectionate memory to the late DANIEL HERBERT EFRON, PH.D., M.D. 1913-1972 cherished friend of the author and of the Botanical Museum, a true scientist devoted to the interdisciplinary approach in the advancement of knowledge. A/""'f""'<J liz {i>U<// t~ },~u 0<<J4 ~ If;. r:J~ ~ //"'~uI ~ ()< d~ ~ !dtd't;:..1 "./.u.L .A Vdl0 If;;: ~ '" OU'''-k4 :/" tu-d ''''''"-''t2.. ?,,".jd,~ jft I;ft'- ?_rl; A~~ ~r'4tft,t -5 " q,.,<,4 ~~ l' #- /""/) -/~ "1'Ii;. ,1""", "/'/1'1",, I X C"'-r'fttt. #) (../..d ~;, . W,( ~ ~ f;r"'" y it;.,,J 11/" Y 4J.. %~~ l{jr~ t> ~~ ~txh '1'ix r 4 6~" c/<'T'''(''-;{' rn« ?.d ~;;1';;/ a-.d txZ-~ ~ ;o/~ <A.H-iz "" ~".,/( 1-/X< "..< ,:" -.... ~ ~ . JJlr-0? on . .it-(,0.1' r 4 -11<.1.- aw./{') -:JL. P7t;;"j~;1 S .d-At ;0~/lAQ<..t ,ti~?,f,.... vj "7rU<-'- ~I""" =iiR-I1;M~ a....k«<-l, ¢- f!!) d..;.:~ M ~ ~y£/1 ~/.u..-... It'--, "" # :Z:-,k. "i ~ "d/~ efL<.<~/ ,w 1'#,') /';~~;d-t a;.. tlArl-<7'" I .Ii;'~.1 (1(-;.,} >Lc -(l"7C),.,..,;.. :.... ,,:/ ~ /-V,~ , ,1" # (i F'"' l' fJ~~A- (.tG- ~/~ Z:--7Co- ,,:. ,L7r= f,-, , ~t) ahd-p;: fJ~ / tr>d .4 ~f- $. b".,,1 ~/. ~ pd. 1'7'-· X ~-t;;;;.,~;z jM ~0Y:tJ;; ~ """.,4? br;K,' ./.n.u" ~ 7r .".,.~,j~ ;;f;tT ~ ..4'./ ;pf,., tJd~ M_~ (./I<'/~.'. IU. et. c./,. ~L.y !f-t.<H>:t;.tu ~ ~,:,-,p., .....:. -

Different Mechanisms Drive the Performance of Native and Invasive Woody Species in Response to Leaf Phosphorus Supply During Periods of Drought Stress and Recovery
Plant Physiology and Biochemistry 82 (2014) 66e75 Contents lists available at ScienceDirect Plant Physiology and Biochemistry journal homepage: www.elsevier.com/locate/plaphy Research article Different mechanisms drive the performance of native and invasive woody species in response to leaf phosphorus supply during periods of drought stress and recovery * Marciel Teixeira Oliveira, Camila Dias Medeiros, Gabriella Frosi, Mauro Guida Santos Departamento de Botanica,^ Laboratorio de Ecofisiologia Vegetal, Universidade Federal de Pernambuco, Recife PE 50670-901, Brasil article info abstract Article history: The effects of drought stress and leaf phosphorus (Pi) supply on photosynthetic metabolism in woody Received 31 March 2014 tropical species are not known, and given the recent global environmental change models that forecast Accepted 13 May 2014 lower precipitation rates and periods of prolonged drought in tropical areas, this type of study is Available online 22 May 2014 increasingly important. The effects of controlled drought stress and Pi supply on potted young plants of two woody species, Anadenanthera colubrina (native) and Prosopis juliflora (invasive), were determined Keywords: by analyzing leaf photosynthetic metabolism, biochemical properties and water potential. In the Chlorophyll fluorescence maximum stress, both species showed higher leaf water potential (Jl) in the treatment drought þPi Climate change À Drought tolerance when compared with the respective control Pi. The native species showed higher gas exchange under þ e Gas exchange drought Pi than under drought Pi conditions, while the invasive species showed the same values Water deficit between drought þPi and ÀPi. Drought affected the photochemical part of photosynthetic machinery more in the invasive species than in the native species. -

A Guide to Lesser Known Tropical Timber Species July 2013 Annual Repo Rt 2012 1 Wwf/Gftn Guide to Lesser Known Tropical Timber Species
A GUIDE TO LESSER KNOWN TROPICAL TIMBER SPECIES JULY 2013 ANNUAL REPO RT 2012 1 WWF/GFTN GUIDE TO LESSER KNOWN TROPICAL TIMBER SPECIES BACKGROUND: BACKGROUND: The heavy exploitation of a few commercially valuable timber species such as Harvesting and sourcing a wider portfolio of species, including LKTS would help Mahogany (Swietenia spp.), Afrormosia (Pericopsis elata), Ramin (Gonostylus relieve pressure on the traditionally harvested and heavily exploited species. spp.), Meranti (Shorea spp.) and Rosewood (Dalbergia spp.), due in major part The use of LKTS, in combination with both FSC certification, and access to high to the insatiable demand from consumer markets, has meant that many species value export markets, could help make sustainable forest management a more are now threatened with extinction. This has led to many of the tropical forests viable alternative in many of WWF’s priority places. being plundered for these highly prized species. Even in forests where there are good levels of forest management, there is a risk of a shift in species composition Markets are hard to change, as buyers from consumer countries often aren’t in natural forest stands. This over-exploitation can also dissuade many forest willing to switch from purchasing the traditional species which they know do managers from obtaining Forest Stewardship Council (FSC) certification for the job for the products that they are used in, and for which there is already their concessions, as many of these high value species are rarely available in a healthy market. To enable the market for LKTS, there is an urgent need to sufficient quantity to cover all of the associated costs of certification. -

Inner Visions: Sacred Plants, Art and Spirituality
AM 9:31 2 12/10/14 2 224926_Covers_DEC10.indd INNER VISIONS: SACRED PLANTS, ART AND SPIRITUALITY Brauer Museum of Art • Valparaiso University Vision 12: Three Types of Sorcerers Gouache on paper, 12 x 16 inches. 1989 Pablo Amaringo 224926_Covers_DEC10.indd 3 12/10/14 9:31 AM 3 224926_Text_Dec12.indd 3 12/12/14 11:42 AM Inner Visions: Sacred Plants, Art and Spirituality • An Exhibition of Art Presented by the Brauer Museum • Curated by Luis Eduardo Luna 4 224926_Text.indd 4 12/9/14 10:00 PM Contents 6 From the Director Gregg Hertzlieb 9 Introduction Robert Sirko 13 Inner Visions: Sacred Plants, Art and Spirituality Luis Eduardo Luna 29 Encountering Other Worlds, Amazonian and Biblical Richard E. DeMaris 35 The Artist and the Shaman: Seen and Unseen Worlds Robert Sirko 73 Exhibition Listing 5 224926_Text.indd 5 12/9/14 10:00 PM From the Director In this Brauer Museum of Art exhibition and accompanying other than earthly existence. Additionally, while some objects publication, expertly curated by the noted scholar Luis Eduardo may be culture specific in their references and nature, they are Luna, we explore the complex and enigmatic topic of the also broadly influential on many levels to, say, contemporary ritual use of sacred plants to achieve visionary states of mind. American and European subcultures, as well as to contemporary Working as a team, Luna, Valparaiso University Associate artistic practices in general. Professor of Art Robert Sirko, Valparaiso University Professor We at the Brauer Museum of Art wish to thank the Richard E. DeMaris and the Brauer Museum staff present following individuals and agencies for making this exhibition our efforts of examining visual products arising from the possible: the Brauer Museum of Art’s Brauer Endowment, ingestion of these sacred plants and brews such as ayahuasca. -

Morphological and Physiological Germination Aspects of Anadenanthera Colubrina (Vell.) Brenan
Available online: www.notulaebotanicae.ro Print ISSN 0255-965X; Electronic 1842-4309 Notulae Botanicae Horti AcademicPres Not Bot Horti Agrobo, 2018, 46(2):593-600. DOI:10.15835/nbha46211094 Agrobotanici Cluj-Napoca Original Article Morphological and Physiological Germination Aspects of Anadenanthera colubrina (Vell.) Brenan Fernanda Carlota NERY 1*, Marcela Carlota NERY 2, Débora de Oliveira PRUDENTE 3, Amauri Alves de ALVARENGA 3, Renato PAIVA 3 1Universidade Federal de São João Del Rei (UFSJ), Departamento de Engenharia de Biossistemas, São João Del Rei - MG, Brazil; [email protected] (*corresponding author) 2Universidade dos Vales do Jequitinhonha e Mucuri (UFVJM), Diamantina – MG, Brazil; [email protected] 3Universidade Federal de Lavras (UFLA), Programa de Pós-graduação em Fisiologia Vegetal, Departamento de Biologia, Lavras – MG, Brazil; [email protected] ; [email protected] ; [email protected] Abstra ct Anadenanthera colubrina is a species native to Brazil, from the Fabaceae family and has potential for use in the timber industry, in the reforestation of degraded areas, besides having medicinal properties. Its propagation is mainly by seeds, but basic subsidies regarding the requirements for optimal germination conditions are still lacking. Aiming to contribute to the expansion of its cultivation, rational use and conservation, the objective of this study was to investigate the morphology and anatomy of fruits and seeds, as well as the responses to factors as thermal regimes and substrates in seed germination. The 1000-seed weight and seeds per fruit were determined. To characterize the seed tissues, histochemical test with Sudan III and Lugol was used. The temperatures analyzed in the germination test were 15-25 °C; 25 °C; 20-30 °C and 30 °C. -

Depicting the Mating System and Patterns of Contemporary Pollen flow in Trees of the Genus Anadenanthera (Fabaceae)
Depicting the mating system and patterns of contemporary pollen flow in trees of the genus Anadenanthera (Fabaceae) Juliana Massimino Feres1,*, Alison G. Nazareno2,*, Leonardo M. Borges3, Marcela Corbo Guidugli1, Fernando Bonifacio-Anacleto1 and Ana Lilia Alzate-Marin1 1 Programa de Pós-Graduação em Genética, Departamento de Genética, Universidade de São Paulo, Faculdade de Medicina de Ribeirão Preto, Ribeirão Preto, SP, Brazil 2 Departamento de Genética, Ecologia e Evolução, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil 3 Departamento de Botânica, Universidade Federal de São Carlos, São Carlos, SP, Brazil * These authors contributed equally to this work. ABSTRACT Anadenanthera (Fabaceae) is endemic to the Neotropics and consists of two tree species: A. colubrina (Vell.) Brenan and A. peregrina (L.) Speg. This study examined the mating system and contemporary gene flow of A. colubrina (Acol) and A. peregrina (Aper) in a highly fragmented area of the Atlantic Forest to provide valuable information that informs conservation strategies. Reproductive adults from forest remnants [nA. colubrina = 30 (2.7 ha), nA. peregrina = 55 (4.0 ha)] and progeny-arrays (nA. colubrina = 322, nA. peregrina = 300) were genotyped for seven nuclear microsatellite markers. Mating system analyses revealed that A. colubrina is a mixed mating species (tm = 0.619) while A. peregrina is a predominantly outcrossing species (tm = 0.905). For both Anadenanthera species, high indices of biparental inbreeding were observed (Acol = 0.159, Aper = 0.216), resulting in low effective pollination neighborhood sizes. Categorical paternity analysis revealed different Submitted 14 July 2020 scales of pollen dispersal distance: the majority of crossings occurring locally Accepted 24 November 2020 (i.e., between nearby trees within the same population), with moderate pollen Published 7 April 2021 dispersal coming from outside the forest fragments boundaries (Acolmp = 30%, Corresponding authors Aper = 35%). -
![IVEW3ITY NO. CXCIII a TAXONO]L'lic STUDY of the GE!\CS](https://docslib.b-cdn.net/cover/1498/ivew3ity-no-cxciii-a-taxono-llic-study-of-the-ge-cs-1501498.webp)
IVEW3ITY NO. CXCIII a TAXONO]L'lic STUDY of the GE!\CS
COi\TI:II)UTIONS FI:O~l THEGI:AY II ERB .~ R lml Of IlAl:VAltD U;\IV EW3ITY J::. lilt".1 b~· lh't',l c. Hl'Jl!i n ~ ull.I Hobtc n c. !-"ht, ~ NO. CXCIII A TAXONO]l'lIC STUDY OF THE GE!\CS ANADE Ai:'i THERA By S ItU Y01'\ REI S ALTSCHl"L Il\FRASPECIFIC VARIATION IN THE GUTlERREZIA SAROTHRAE CmlPLEX (COMPOSITAE-ASTEEEAE) By Orro T. SOLBRIG Published by THE GR AY HERBARIUlII OF HARVARD UKIVERSITY CAMBRIDGE, MASS., U. S. A. , MANUFACTURED BY THE LEXINGTON PRESS, INC. LEXINGTON. MASSACHUSETTS A TAX O:\O,llC STUDY OF TilE GE~ljS AN A DENA:\TI-IEI~A SIR! VO:-i HE!S ALT~l-HUL The "nail, l ropical to suutropical, and strictly :\ew World genu s .AlIlLl!C"1Wllth c,'n formerly was I...'ons ide}' ~d n~ $~c tion NioJ)(I of the genus Pip to.(/ cllia and i$ the cor.',m on It.'gumi 11 0 U S sou rce of lhe so-called narcotics known as COIIO/>O, l-iica a nd Yopo, Because of lheir ullusual c"rects upon th~ human nervollS system, the chemical constitutt.'nts of t~.ese material s are of cUlTen t interest in the field of ~xper im " nta l psychia lry, Among the chemical compound, which leave u.een iso lated from the species of A,wde1U",th ,Ta, may be included some hallucinogenic cl!'ugs designated as psychotomimetics (Hofmann, 1959) , In contrast to otlwr psychotropic drugs, which merely evoke mod ifications in the mood of th~ reci pi ent, the psychotomimetics appeal' to produce profound and acute changes in perception_ An interest both in plants of possibl~ medical significance and in species in need of t"xonomic stud,' provided a stimulus for the research whose results are published in part in this paper. -

Seed Germination and Seedling Development of Anadenanthera Colubrina in Response to Weight and Temperature Conditions
Journal of Pla nt Sciences 2014; 2(1): 37-42 Published online February 20, 2014 (http://www.sciencepublishinggroup.com/j/jps) doi: 10.11648/j.jps.20140201.17 Seed germination and seedling development of Anadenanthera colubrina in response to weight and temperature conditions Francisco Carlos Barboza Nogueira 1, *, Charles Lobo Pinheiro 1, Sebastião Medeiros Filho 1, Dalva Maria da Silva Matos 2 1Departamento de Fitotecnica, Universidade Federal do Ceará - UFC, Campus do Pici Fortaleza, Ceará, Brazil 2Departamento de Hidrobiologia, Universidade Federal de São Carlos, via Washington Luis, São Carlos, São Paulo, Brazil Email address: [email protected] (F. C. B. Nogueira) To cite this article: Francisco Carlos Barboza Nogueira, Charles Lobo Pinheiro, Sebastião Medeiros Filho, Dalva Maria da Silva Matos. Seed Germination and Seedling Development of Anadenanthera Colubrina in Response to Weight and Temperature Conditions. Journal of Plant Sciences. Vol. 2, No. 1, 2014, pp. 37-42. doi: 10.11648/j.jps.20140201.17 Abstract: The aim of the study was investigated the effects of seed weight and temperature on the germination behavior, and development of seedlings of Anadenanthera colubrina . Germination was carried out using the five constant temperature regimes (20, 25, 30, 35 and 40ºC) under photoperiod (12h) and two classes of seeds: heavy (≥ 0.095g) and light (<0.095g). The experimental design was totally random, with four replications of 25 seeds. The temperature was significant for germination percentage, germination velocity index and mean germination time. There was no effect of seed weight, and there was no interaction between temperature and seed weight for three response variables. Optimum germination for A. -

Size and Vigor of Anadenanthera Colubrina (Vell.) Brenan Seeds Harvested in Caatinga Areas1
Journal of Seed Science, v.39, n.4, p.363-373, 2017 http://dx.doi.org/10.1590/2317-1545v39n4173727 Size and vigor of Anadenanthera colubrina (Vell.) Brenan seeds harvested in Caatinga areas1 Jaciara de Souza Bispo2, Danielle Carolina Campos da Costa2, Samara Elizabeth Vieira Gomes2, Gilmara Moreira de Oliveira3, Janete Rodrigues Matias4, Renata Conduru Ribeiro5, Bárbara França Dantas5* ABSTRACT - Angico is a species found in several environments in Brazil, with several applications. It is used in the timber industry and mainly in folk medicine. In order to verify a variation in the biometric characteristics and the quality of seeds from different mother-plants in different harvesting years, the following variables were studied: moisture content, diameter, density, electrical conductivity, fresh and dry matter of seedlings, germination percentage and kinetics, in a completely randomized design with a 2x3 factorial arrangement (lots x size). The obtained results showed that angico seeds from different lots showed different physiological quality, possibly due to the climate variations to which mother-plants were submitted in the different years. Seed size directly interferes with seedling growth under both controlled and greenhouse conditions, and it can be used as a vigor indicator for angico seeds. Index terms: physiological quality, biometrics, Fabaceae, angico. Tamanho e vigor de sementes de Anadenanthera colubrina (Vell.) Brenan colhidas em área de Caatinga RESUMO - O angico é uma espécie encontrada em diversos ambientes no Brasil, -

Silviculture in the Tropics (Tropical Forestry)
Tropical Forestry For other volumes of the series, go to www.springer.com/series/5439 . Sven Gu¨nter l Michael Weber l Bernd Stimm l Reinhard Mosandl Editors Silviculture in the Tropics With 49 Figures and 59 Tables Editors PD Dr. Sven Gu¨nter Prof. Dr. Michael Weber Dr. Bernd Stimm Prof. Dr. Reinhard Mosandl Institute of Silviculture Center of Life and Food Sciences Weihenstephan Technische Universita¨t Mu¨nchen Hans-Carl-von-Carlowitz-Platz 2 85354 Freising Germany [email protected] [email protected] [email protected] [email protected] ISSN 1614-9785 ISBN 978-3-642-19985-1 e-ISBN 978-3-642-19986-8 DOI 10.1007/978-3-642-19986-8 Springer Heidelberg Dordrecht London New York Library of Congress Control Number: 2011932166 # Springer-Verlag Berlin Heidelberg 2011 This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilm or in any other way, and storage in data banks. Duplication of this publication or parts thereof is permitted only under the provisions of the German Copyright Law of September 9, 1965, in its current version, and permission for use must always be obtained from Springer. Violations are liable to prosecution under the German Copyright Law. The use of general descriptive names, registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protec- tive laws and regulations and therefore free for general use.