Methoxy-6-Methylflavone and Kavain at Recombinant GABAA Receptors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
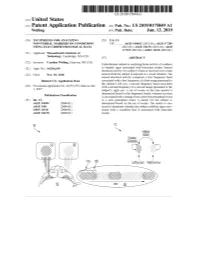
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Highly Water-Soluble Solid Dispersions of Honokiol: Preparation, Solubility, and Bioavailability Studies and Anti-Tumor Activity Evaluation
pharmaceutics Article Highly Water-Soluble Solid Dispersions of Honokiol: Preparation, Solubility, and Bioavailability Studies and Anti-Tumor Activity Evaluation Li Wang 1,2, Weiwei Wu 1,2, Lingling Wang 1,2, Lu Wang 1,2 and Xiuhua Zhao 1,2,* 1 College of Chemistry, Chemical Engineering and Resource Utilization, Northeast Forestry University, Harbin 150040, China; [email protected] (L.W.); [email protected] (W.W.); [email protected] (L.W.); [email protected] (L.W.) 2 Key Laboratory of Forest Plant Ecology, Northeast Forestry University, Ministry of Education, Harbin 150040, China * Correspondence: [email protected]; Tel.: +86-451-82191517; Fax: +86-451-82102082 Received: 18 September 2019; Accepted: 24 October 2019; Published: 1 November 2019 Abstract: Honokiol (HK), a well-tolerated natural product, has many multiple pharmacological activities. However, its poor water solubility and low bioavailability limit its clinical application and development. The aim of this research was to prepare the solid dispersion (SD) formulation of honokiol (HK) with poloxamer-188 (PLX) as the carrier, thereby improving its solubility and oral bioavailability. Firstly, by investigating the relationship between the addition amount of the PLX and the solubility of HK, and the effects of solid dispersions with different ratios of HK–PLX on the solubility of HK, we determined that the optimum ratio of PLX to HK was (1:4). Then, the HK–PLX (1:4) SD of HK was prepared using the solvent evaporation method. The morphology of the obtained HK–PLX (1:4) SD was different from that of free HK. The HK in the HK–PLX (1:4) SD existed in amorphous form and formed intermolecular hydrogen bonds with PLX. -

Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects
Neuropsychopharmacology (2005) 30, 833–841 & 2005 Nature Publishing Group All rights reserved 0893-133X/05 $30.00 www.neuropsychopharmacology.org Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects 1 2 ,3 1 Stefan Mathias , Josef Zihl , Axel Steiger* and Marike Lancel 1Section of Sleep Pharmacology, Max-Planck-Institute of Psychiatry, Munich, Germany; 2Section of Neuropsychology, Max-Planck-Institute of Psychiatry, Munich, Germany; 3Department of Psychiatry, Max-Planck-Institute of Psychiatry, Munich, Germany Aging is associated with dramatic reductions in sleep continuity and sleep intensity. Since gaboxadol, a selective GABAA receptor agonist, has been demonstrated to improve sleep consolidation and promote deep sleep, it may be an effective hypnotic, particularly for elderly patients with insomnia. In the present study, we investigated the effects of subchronic gaboxadol administration on nocturnal sleep and its residual effects during the next days in elderly subjects. This was a randomized, double-blind, placebo-controlled, balanced crossover study in 10 healthy elderly subjects without sleep complaints. The subjects were administered either placebo or 15 mg gaboxadol hydrochloride at bedtime on three consecutive nights. Sleep was recorded during each night from 2300 to 0700 h and tests assessing attention (target detection, stroop test) and memory function (visual form recognition, immediate word recall, digit span) were applied at 0900, 1400, and 1700 h during the following days. Compared with placebo, gaboxadol significantly shortened subjective sleep onset latency and increased self-rated sleep intensity and quality. Polysomnographic recordings showed that it significantly decreased the number of awakenings, the amount of intermittent wakefulness, and stage 1, and increased slow wave sleep and stage 2. -

Valerenic Acid Potentiates and Inhibits GABAA Receptors: Molecular Mechanism and Subunit Specificity
ARTICLE IN PRESS + MODEL Neuropharmacology xx (2007) 1e10 www.elsevier.com/locate/neuropharm Valerenic acid potentiates and inhibits GABAA receptors: Molecular mechanism and subunit specificity S. Khom a, I. Baburin a, E. Timin a, A. Hohaus a, G. Trauner b, B. Kopp b, S. Hering a,* a Department of Pharmacology and Toxicology, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria b Department of Pharmacognosy, University of Vienna, Althanstrasse 14, A-1090 Vienna, Austria Received 8 December 2006; received in revised form 11 April 2007; accepted 30 April 2007 Abstract Valerian is a commonly used herbal medicinal product for the treatment of anxiety and insomnia. Here we report the stimulation of chloride currents through GABAA receptors (IGABA) by valerenic acid (VA), a constituent of Valerian. To analyse the molecular basis of VA action, we expressed GABAA receptors with 13 different subunit compositions in Xenopus oocytes and measured IGABA using the two-microelectrode voltage-clamp technique. We report a subtype-dependent stimulation of IGABA by VA. Only channels incorporating b2 or b3 subunits were stimulated by VA. Replacing b2/3 by b1 drastically reduced the sensitivity of the resulting GABAA channels. The stimulatory effect of VA on a1b2 receptors was substantially reduced by the point mutation b2N265S (known to inhibit loreclezole action). Mutating the corresponding residue of b1 (b1S290N) induced VA sensitivity in a1b1S290N comparable to a1b2 receptors. Modulation of IGABA was not significantly dependent on incorporation of a1, a2, a3 or a5 subunits. VA displayed a significantly lower efficiency on channels incorporating a4 subunits. IGABA modulation by VA was not g subunit dependent and not inhibited by flumazenil (1 mM). -

(12) Patent Application Publication (10) Pub. No.: US 2006/014 1075A1 Talbott (43) Pub
US 2006O141075A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/014 1075A1 Talbott (43) Pub. Date: Jun. 29, 2006 (54) METHODS AND COMPOSITIONS FOR Publication Classification WEIGHT MANAGEMENT AND MOOD ENHANCEMENT (51) Int. Cl. A6IR 36/82 (2006.01) A6IR 36/752 (2006.01) (76) Inventor: Shawn M. Talbott, Draper, UT (US) A6II 3/522 (2006.01) A6II 3/56 (2006.01) Correspondence Address: A6IR 33/26 (2006.01) MACPHERSON KWOK CHEN & HEID LLP A 6LX 36/575 (2006.01) 1762 TECHNOLOGY DRIVE, SUITE 226 (52) U.S. Cl. ......................... 424/729; 424/736: 424/774; SAN JOSE, CA 95110 (US) 424/769; 514/263.31; 514/171; 424/646 (21) Appl. No.: 11/360,312 (57) ABSTRACT The present invention provides nutritional Supplement com (22) Filed: Feb. 23, 2006 positions and processes for treatment using nutritional Supplements that assist in weight management. One aspect Related U.S. Application Data of the invention comprises the inclusion in a single Supple ment constituent(s) that assist in reduction of a subjects (62) Division of application No. 10/895.253, filed on Jul. cortisol level, thermogenic constituent(s), and constituent(s) 20, 2004. that assist in stabilizing blood Sugar levels. Patent Application Publication Jun. 29, 2006 Sheet 1 of 4 US 2006/01.41075A1 Global Mood State (Profile of Mood States) 160 -8.6% 150 140 130 120 o Mood, Pre 110 Effect of a stress/cortisol-controlling dietary supplement on Global Mood States following 12 weeks of supplementation. P = placebo. S = Supplement. * = P <0.05 compared to Pre. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Anxiety Disorders and GABA Neurotransmission: a Disturbance of Modulation
Journal name: Neuropsychiatric Disease and Treatment Article Designation: REVIEW Year: 2015 Volume: 11 Neuropsychiatric Disease and Treatment Dovepress Running head verso: Nuss Running head recto: Anxiety and modulation open access to scientific and medical research DOI: http://dx.doi.org/10.2147/NDT.S58841 Open Access Full Text Article REVIEW Anxiety disorders and GABA neurotransmission: a disturbance of modulation Philippe Nuss1,2 Abstract: Lines of evidence coming from many branches of neuroscience indicate that anxiety 1Department of Psychiatry, Hôpital St disorders arise from a dysfunction in the modulation of brain circuits which regulate emotional Antoine, AP-HP, 2UMR 7203, INSERM responses to potentially threatening stimuli. The concept of anxiety disorders as a disturbance ERL 1057 – Bioactive Molecules of emotional response regulation is a useful one as it allows anxiety to be explained in terms Laboratory, Pierre and Marie Curie University, Paris, France of a more general model of aberrant salience and also because it identifies avenues for devel- oping psychological, behavioral, and pharmacological strategies for the treatment of anxiety disorder. These circuits involve bottom-up activity from the amygdala, indicating the presence of potentially threatening stimuli, and top-down control mechanisms originating in the prefron- tal cortex, signaling the emotional salience of stimuli. Understanding the factors that control cortical mechanisms may open the way to identification of more effective cognitive behavioral strategies for managing anxiety disorders. The brain circuits in the amygdala are thought to For personal use only. comprise inhibitory networks of γ-aminobutyric acid-ergic (GABAergic) interneurons and this neurotransmitter thus plays a key role in the modulation of anxiety responses both in the normal and pathological state. -

Calcium Channel Blocker As a Drug Candidate for the Treatment of Generalised Epilepsies
UNIVERSITAT DE BARCELONA Faculty of Pharmacy and Food Sciences Calcium channel blocker as a drug candidate for the treatment of generalised epilepsies Final degree project Author: Janire Sanz Sevilla Bachelor's degree in Pharmacy Primary field: Organic Chemistry, Pharmacology and Therapeutics Secondary field: Physiology, Pathophysiology and Molecular Biology March 2019 This work is licensed under a Creative Commons license ABBREVIATIONS AED antiepileptic drug AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid ANNA-1 antineuronal nuclear antibody 1 BBB blood-brain barrier Bn benzyl BnBr benzyl bromide BnNCO benzyl isocyanate Boc tert-butoxycarbonyl Bu4NBr tetrabutylammonium bromide Ca+2 calcium ion CACNA1 calcium channel voltage-dependent gene cAMP cyclic adenosine monophosphate CCB calcium channel blocker cGMP cyclic guanosine monophosphate CH3CN acetonitrile Cl- chlorine ion Cmax maximum concentration CMV cytomegalovirus CTScan computed axial tomography DCM dichloromethane DIPEA N,N-diisopropylethylamine DMF dimethylformamide DMPK drug metabolism and pharmacokinetics DNET dysembryoplastic neuroepithelial tumours EEG electroencephalogram EPSP excitatory post-synaptic potential FDA food and drug administration Fe iron FLIPR fluorescence imaging plate reader fMRI functional magnetic resonance imaging GABA γ-amino-α-hydroxybutyric acid GAD65 glutamic acid decarboxylase 65 GAERS generalised absence epilepsy rat of Strasbourg GluR5 kainate receptor GTC generalised tonic-clonic H+ hydrogen ion H2 hydrogen H2O dihydrogen dioxide (water) -

Herbal Insomnia Medications That Target Gabaergic Systems: a Review of the Psychopharmacological Evidence
Send Orders for Reprints to [email protected] Current Neuropharmacology, 2014, 12, 000-000 1 Herbal Insomnia Medications that Target GABAergic Systems: A Review of the Psychopharmacological Evidence Yuan Shia, Jing-Wen Donga, Jiang-He Zhaob, Li-Na Tanga and Jian-Jun Zhanga,* aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, P.R. China; bDepartment of Pharmacology, School of Marine, Shandong University, Weihai, P.R. China Abstract: Insomnia is a common sleep disorder which is prevalent in women and the elderly. Current insomnia drugs mainly target the -aminobutyric acid (GABA) receptor, melatonin receptor, histamine receptor, orexin, and serotonin receptor. GABAA receptor modulators are ordinarily used to manage insomnia, but they are known to affect sleep maintenance, including residual effects, tolerance, and dependence. In an effort to discover new drugs that relieve insomnia symptoms while avoiding side effects, numerous studies focusing on the neurotransmitter GABA and herbal medicines have been conducted. Traditional herbal medicines, such as Piper methysticum and the seed of Zizyphus jujuba Mill var. spinosa, have been widely reported to improve sleep and other mental disorders. These herbal medicines have been applied for many years in folk medicine, and extracts of these medicines have been used to study their pharmacological actions and mechanisms. Although effective and relatively safe, natural plant products have some side effects, such as hepatotoxicity and skin reactions effects of Piper methysticum. In addition, there are insufficient evidences to certify the safety of most traditional herbal medicine. In this review, we provide an overview of the current state of knowledge regarding a variety of natural plant products that are commonly used to treat insomnia to facilitate future studies. -

Central Valley Toxicology Drug List
Chloroform ~F~ Lithium ~A~ Chlorpheniramine Loratadine Famotidine Acebutolol Chlorpromazine Lorazepam Fenoprofen Acetaminophen Cimetidine Loxapine Fentanyl Acetone Citalopram LSD (Lysergide) Fexofenadine 6-mono- Clomipramine acetylmorphine Flecainide ~M~ Clonazepam a-Hydroxyalprazolam Fluconazole Maprotiline Clonidine a-Hydroxytriazolam Flunitrazepam MDA Clorazepate Albuterol Fluoxetine MDMA Clozapine Alprazolam Fluphenazine Medazepam Cocaethylene Amantadine Flurazepam Meperidine Cocaine 7-Aminoflunitrazepam Fluvoxamine Mephobarbital Codeine Amiodarone Fosinopril Meprobamate Conine Amitriptyline Furosemide Mesoridazine Cotinine Amlodipine Methadone Cyanide ~G~ Amobarbital Methanol Cyclobenzaprine Gabapentin Amoxapine d-Methamphetamine Cyclosporine GHB d-Amphetamine l-Methamphetamine Glutethamide l-Amphetamine ~D~ Methapyrilene Guaifenesin Aprobarbital Demoxepam Methaqualone Atenolol Desalkylfurazepam ~H~ Methocarbamol Atropine Desipramine Halazepam Methylphenidate ~B~ Desmethyldoxepin Haloperidol Methyprylon Dextromethoraphan Heroin Metoclopramide Baclofen Diazepam Hexobarbital Metoprolol Barbital Digoxin Hydrocodone Mexiletine Benzoylecgonine Dihydrocodein Hydromorphone Midazolam Benzphetamine Dihydrokevain Hydroxychloroquine Mirtazapine Benztropine Diltiazem Hydroxyzine Morphine (Total/Free) Brodificoum Dimenhydrinate Bromazepam ~N~ Diphenhydramine ~I~ Bupivacaine Nafcillin Disopyramide Ibuprofen Buprenorphine Naloxone Doxapram Imipramine Bupropion Naltrexone Doxazosin Indomethacin Buspirone NAPA Doxepin Isoniazid Butabarbital Naproxen -

Molecular Mechanisms Driving Prostate Cancer Neuroendocrine Differentiation
Molecular mechanisms driving prostate cancer neuroendocrine differentiation Submitted by Joseph Edward Sutton Supervisory team: Dr Amy Poole (DoS) Dr Jennifer Fraser Dr Gary Hutchison A thesis submitted in partial fulfilment of the requirements of Edinburgh Napier University, for the award of Doctor of Philosophy. October 2019 School of Applied Sciences Edinburgh Napier University Edinburgh Declaration It is hereby declared that this thesis is the result of the author’s original research. It has been composed by the author and has not been previously submitted for examination which has led to the award of a degree. Signed: II Dedication This thesis is dedicated to my grandfather William ‘Harry’ Russell, who died of stomach cancer in 2014. Thank you for always encouraging me to achieve my ambitions, believing in me and for retaining your incredible positivity and sense of humour, even at the very end of your life. III Acknowledgements First of all, I would like to acknowledge my parents, who dedicated so much effort and energy into helping me to achieve my lifelong ambition of becoming a scientist. From taking me to the Natural History and Science Museums in London as a child, to tolerating my obsession with Jurassic Park and continuing to support me in both of your unique yet equally important ways, thank you. I would also like to thank my PhD supervisors Dr Amy Poole and Dr Jenny Fraser, not only for their excellent scientific guidance but also for their great banter and encouragement along the way. Thank you for seeing some potential in me, taking a chance on me and for helping me to continue my scientific journey. -

85 Mv Respectively. When the Membranewas Depolarized, Th
J. Physiol. (1985), 360, pp. 161-185 161 With 14 text-figurem Printed in Great Britain COMPARISON OF THE ACTION OF BACLOFEN WITH y-AMINOBUTYRIC ACID ON RAT HIPPOCAMPAL PYRAMIDAL CELLS IN VITRO BY N. R. NEWBERRY* AND R. A. NICOLLt From the Departments of Pharmacology and Physiology, University of California, San Francisco, CA 94143, U.S.A. (Received 13 June 1984) SUMMARY 1. Intracellular recordings from CAI pyramidal cells in the hippocampal slice preparation were used to compare the action of baclofen, a y-aminobutyric acid (GABA) analogue, with GABA. 2. Ionophoretic application of GABA or baclofen into stratum (s.) pyramidale evoked hyperpolarizations associated with reductions in the input resistance of the cell. Baclofen responses were easier to elicit in the dendrites than in the cell body layer. 3. Blockade of synaptic transmission, with tetrodotoxin or cadmium, did not reduce baclofen responses, indicating a direct post-synaptic action. 4. (+ )-Bicuculline (10 ,UM) and bicuculline methiodide (100 /SM) had little effect on baclofen responses but strongly antagonized somatic GABA responses of equal amplitude. The bicuculline resistance of the baclofen response was not absolute, as higher concentrations of these compounds did reduce it. Pentobarbitone (100 /M) enhanced somatic GABA responses without affecting baclofen responses. (-)-Baclofen was approximately 200 times more potent than (+ )-baclofen. 5. The reversal potentials for the somatic GABA and baclofen responses were -70 mV and -85 mV respectively. When the membrane was depolarized, the baclofen response was reduced. This apparent voltage sensitivity was not seen with somatic GABA responses. 6. Altering the chloride gradient across the cell membrane altered the reversal potential of the somatic GABA response but not that ofthe baclofen response.