Some Contributions to Deep Learning for Metagenomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

General Agreement on Tariffs and Trade Accord
GENERAL AGREEMENT ON TARIFFS AND TRADE MIN(86)/INF/3 ACCORD GENERAL SUR LES TARIFS DOUANIERS ET LE COMMERCE 17 September 1986 ACUERDO GENERAL SOBRE ARANCELES ADUANEROS Y COMERCIO Limited Distribution CONTRACTING PARTIES PARTIES CONTRACTANTES PARTES CONTRATANTIS Session at Ministerial Session à l'échelon Periodo de sesiones a nivel Level ministériel ministerial 15-19 September 1986 15-19 septembre 1986 15-19 setiembre 1986 LIST OF REPRESENTATIVES LISTE DES REPRESENTANTS LISTA DE REPRESENTANTES Chairman: S.E. Sr. Enrique Iglesias Président; Ministro de Relaciones Exteriores Présidente; de la Republica Oriental del Uruguay ARGENTINA Représentantes Lie. Dante Caputo Ministro de Relaciones Exteriores y Culto » Dr. Juan V. Sourrouille Ministro de Economia Dr. Roberto Lavagna Secretario de Industria y Comercio Exterior Ing. Lucio Reca Secretario de Agricultura, Ganaderïa y Pesca Dr. Bernardo Grinspun Secretario de Planificaciôn Dr. Adolfo Canitrot Secretario de Coordinaciôn Econômica 86-1560 MIN(86)/INF/3 Page 2 ARGENTINA (cont) Représentantes (cont) S.E. Sr. Jorge Romero Embajador Subsecretario de Relaciones Internacionales Econômicas Lie. Guillermo Campbell Subsecretario de Intercambio Comercial Dr. Marcelo Kiguel Vicepresidente del Banco Central de la Republica Argentina S.E. Leopoldo Tettamanti Embaj ador Représentante Permanante ante la Oficina de las Naciones Unidas en Ginebra S.E. Carlos H. Perette Embajador Représentante Permanente de la Republica Argentina ante la Republica Oriental del Uruguay S.E. Ricardo Campero Embaj ador Représentante Permanente de la Republica Argentina ante la ALADI Sr. Pablo Quiroga Secretario Ejecutivo del Comité de Politicas de Exportaciones Dr. Jorge Cort Présidente de la Junta Nacional de Granos Sr. Emilio Ramôn Pardo Ministro Plenipotenciario Director de Relaciones Econômicas Multilatérales del Ministerio de Relaciones Exteriores y Culto Sr. -

La France Après Charlie : Quelques Questions Sensibles, Parmi D'autres, À Mettre En Débat
La France après Charlie : quelques questions sensibles, parmi d’autres, à mettre en débat (Jean-Claude SOMMAIRE 23 03 2015) Une onde de choc de forte amplitude Les tueries de ce début d’année, à Charlie Hebdo et à l’Hyper Cacher de la Porte de Vincennes, ont produit une onde de choc de très forte amplitude sur l’opinion publique de notre pays. On a pu en mesurer l’effet, dès le week-end suivant, quand des marches républicaines, d’une ampleur inattendue, se sont déroulé un peu partout en France, à Paris comme en province. La force de ce séisme, sur nos compatriotes, peut s’expliquer par la conjonction d’un ensemble d’évènements exceptionnels : l’exécution à la kalachnikov, sur leur lieu de travail, d’une équipe de journalistes et de dessinateurs en raison de leur impertinence à l’égard de l’islam, la prise en otage et l’assassinat de clients d’un magasin cacher au seul motif qu’ils étaient juifs, l’achèvement, à terre, d’un policier blessé d’origine maghrébine, etc. Cependant, pour beaucoup d’entre nous, l’élément le plus déstabilisant de ces journées tragiques aura été que ces meurtriers fanatiques ne venaient pas d’une contrée lointaine, où la sauvagerie règne de façon habituelle. Les frères Kouachi et Amedy Coulibaly, nés et scolarisés en France, vivaient près de chez nous, ils étaient nos compatriotes et nos voisins. Comme on avait commencé à le pressentir avec Mohamed Merah, ce trio sanguinaire nous a démontré que l’école laïque, les dispositifs sociaux de l’Etat providence, et les valeurs de la République, ne faisaient pas obstacle au développement de la barbarie islamiste dans nos banlieues. -

Program & Participants List
Bringing people together to accelerate growth and well-being in emerging markets Program and Participants List INAUGURAL MEETING OF AFRICA EMERGING MARKETS FORUM SEPTEMBER 30 - OCTOBER 1, 2007 GERZENSEE, SWITZERLAND Growth and Development in Emerging Market Countries THEEMERGINGMARKETSFORUM INAUGURAL MEETING AFRICA EMERGING MARKETS FORUM GERZENSEE, SWITZERLAND SEPTEMBER 30 - OCTOBER 1, 2007 CONTENTS WELCOME ................................................................................... 3 PROGRAM ................................................................................... 4 BACKGROUND PAPER ................................................................ 5 PROPOSED STEERING COMMITTEE FOR AFRICA ................ 11 LIST OF PARTICIPANTS ........................................................... 12 PARTICIPANTS’ PROFILE ......................................................... 14 HELPFUL INFORMATION ......................................................... 43 THEEMERGINGMARKETSFORUM 2 AFRICA EMERGING MARKETS FORUM THEEMERGINGMARKETSFORUM WELCOME September, 20, 2007 All of us at the Emerging Markets Forum are delighted that you are joining us in Gerzensee at the Inaugural Meeting of the Emerging Markets Forum for Africa. I thank you in advance for your participation and valuable contribution. After our Global Meetings in Oxford in 2005 and in Jakarta 2006, this new initiative on Africa will allow us to look deeper into the economic and social challenges facing the African continent, and also reinforce the Forum’s role as a bridge -

Yale University Library Digital Repository Contact Information
Yale University Library Digital Repository Collection Name: Henry A. Kissinger papers, part II Series Title: Series III. Post-Government Career Box: 723 Folder: 9 Folder Title: Interview about François Mitterrand, Nouvel Observateur, May 24, 1995 Persistent URL: http://yul-fi-prd1.library.yale.internal/catalog/digcoll:558814 Repository: Manuscripts and Archives, Yale University Library Contact Information Phone: (203) 432-1735 Email: [email protected] Mail: Manuscripts and Archives Sterling Memorial Library Sterling Memorial Library P.O. Box 208240 New Haven, CT 06520 Your use of Yale University Library Digital Repository indicates your acceptance of the Terms & Conditions of Use http://guides.library.yale.edu/about/policies/copyright Find additional works at: http://yul-fi-prd1.library.yale.internal GLe-3 MAX ARMANET ReDACTEUR EN CHEF Olifervateur 10-12. PLACE DE LA BOURSE TEL (1) 44.88.34.1 2 78081 PARIS CEDEX 02 FAX (1) 44.08.37.82 te ritatetitl 4 MITTI Eiri"_Ih A N D 50 pages de témoignages pour l'Histoire Robert Badinter Tahar Ben Jelloun George Bush Jean Daniel Jacques Delors Françoise Girond MikhaY1 Gorbatchev Jacques Julliani Henry Kissinger Jean Lacouture Emmanuel I,e Roy Ladurie Pierre Mauroy Jean d'Ormesson Shimon Peres Claude Roy Mario Soares Philippe Sollers Alain Muraine Michel Tournier M 2228- 1593- 20,00 F 2000 CFA ZONE N i593 DU 18 AU 24 MAI 1995 120 FB 510 ES. AILE 8 DM CAN $ 4.2s.430 PTAS 8000 LIN RCI SUAI 2000 CFA CFA 2000 MAROC 22 DU TUNIS 1,8 DTU ANTIL L ES NEUN 22,50 I ()SA NY 3 98 09 E 220 vat vals [rD [atm [go Mon jugement sur la Pyramide avait évolué comme mon sentiment à son égard :je n'aimais pas l'idée au départ maisj'ai été séduit par son exécution Par Henry e Nouvel Observateur. -

2019 Fall Florida International University Commencement
Florida International University FIU Digital Commons FIU Commencement Programs Special Collections and University Archives Fall 2019 2019 Fall Florida International University Commencement Florida International University Follow this and additional works at: https://digitalcommons.fiu.edu/commencement_programs Recommended Citation Florida International University, "2019 Fall Florida International University Commencement" (2019). FIU Commencement Programs. 2. https://digitalcommons.fiu.edu/commencement_programs/2 This work is brought to you for free and open access by the Special Collections and University Archives at FIU Digital Commons. It has been accepted for inclusion in FIU Commencement Programs by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. Florida International University Ocean Bank Convocation Center CommencementModesto A. Maidique Campus, Miami, Florida Saturday, December 14, 2019 Sunday, December 15, 2019 Monday, December 16, 2019 Tuesday, December 17, 2019 Table of Contents Order of Commencement Ceremonies ........................................................................................................3 An Academic Tradition ..............................................................................................................................18 University Governance and Administration ...............................................................................................19 The Honors College ...................................................................................................................................22 -

Dialogue Between Peoples and Cultures in the Euro-Mediterranean Area
Report by the High-Level Advisory Group established at the initiative of the President of the European Commission Courtesy of Fondazione Laboratorio Mediterraneo* Dialogue Between Peoples and Cultures in the Euro-Mediterranean Area Co-Chairs of the Group: Assia Alaoui Bensalah Jean Daniel Members of the Group: Malek Chebel, Juan Diez Nicolas, Umberto Eco, Shmuel N. Eisenstadt, George Joffé, Ahmed Kamal Aboulmagd, Bichara Khader, Adnan Wafic Kassar, Pedrag Matvejevic, Rostane Mehdi, Fatima Mernissi, Tariq Ramadan, Faruk Sen, Faouzi Skali, Simone Susskind-Weinberger and Tullia Zevi This Report represents the opinion of the High-Level Advisory Group only and does not necessarily reflect the views of the European Commission. * The orientation of this map corresponds to the world view of the Arab cartographers of the Middle Ages. Brussels, October 2003 Version DEF Report by the High-Level Advisory Group EXECUTIVE SUMMARY It is difficult to consider the Mediterranean as a coherent whole without taking account of the fractures which divide it, the conflicts which are tearing it apart: Palestine-Israel, Lebanon, Cyprus, the western Balkans, Greece-Turkey, Algeria; incidents with their roots in other, more distant wars such as those in Afghanistan or Iraq, and so on. The Mediterranean is made up of a number of sub-units which challenge or refute unifying ideas. Conflict, however, is not inevitable; it is not its predestined fate. It is this that convinced Romano Prodi, the President of the European Commission, to set up a High- Level Advisory Group. The group set its work on the dialogue between peoples and cultures in the broader context of economic globalisation, enlargement of the European Union, the permanent presence on its soil of communities of immigrant origin, and the questions about identity which these changes are throwing up on both shores of the Mediterranean. -

Download Download
eTropic: electronic journal of studies in the tropics Repurposing, Recycling, Revisioning: Pacific Arts and the (Post)colonial Jean Anderson https://orcid.org/0000-0002-3280-3225 Te Herenga Waka - Victoria University of Wellington, Aotearoa/New Zealand Abstract Taking a broad approach to the concept of recycling, I refer to a range of works, from sculpture to film, street art and poetry, which depict issues of importance to Indigenous peoples faced with the (after) effects of colonisation. Does the use of repurposed materials and/or the knowledge that these objects are the work of Indigenous creators change the way we respond to these works, and if so, how? Keywords: Pacific artists, food colonisation, reappropriation, speaking back, culture eTropic: electronic journal of studies in the tropics publishes new research from arts, humanities, social sciences and allied fields on the variety and interrelatedness of nature, culture, and society in the tropics. Published by James Cook University, a leading research institution on critical issues facing the worlds’ Tropics. Free open access, Scopus, Google Scholar, DOAJ, Crossref, Ulrich's, SHERPA/RoMEO, Pandora, ISSN 1448-2940. Creative Commons CC BY 4.0. Articles are free to download, save and reproduce. Citation: to cite this article include Author(s), title, eTropic, volume, issue, year, pages and DOI: http://dx.doi.org/10.25120/etropic.19.1.2020.3733 eTropic 19.1 (2020) Special Issue: ‘Environmental Artistic Practices and Indigeneity’ 186 Introduction his study takes a broad view of recycling, looking at examples from a range of forms of artistic expression, to explore ways in which Indigenous artists in the T Pacific make use of repurposed materials to criticise some of the profoundly unsettling – and enduring – effects of colonisation. -

The Accuracy Versus Interpretability Trade-Off in Fraud Detection Model
Data & Policy (2021), 3: e12 doi:10.1017/dap.2021.3 RESEARCH ARTICLE The accuracy versus interpretability trade-off in fraud detection model Anna Nesvijevskaia1,2,* , Sophie Ouillade1, Pauline Guilmin1 and Jean-Daniel Zucker3 1Quinten, 8 rue Vernier, Paris 75017, France 2Laboratory DICEN Ile de France, Conservatoire National des Arts et Métiers, 292 rue Saint Martin, Paris 75003, France 3IRD, UMMISCO, Sorbonne University, Bondy F-93143, France *Corresponding author. E-mail: [email protected] Received: 12 October 2020; Revised: 13 March 2021; Accepted: 22 April 2021 Key words: data analysis; fraud detection; human data mediation; interpretability; unbalanced data Abstract Like a hydra, fraudsters adapt and circumvent increasingly sophisticated barriers erected by public or private institutions. Among these institutions, banks must quickly take measures to avoid losses while guaranteeing the satisfaction of law-abiding customers. Facing an expanding flow of operations, effective banking relies on data analytics to support established risk control processes, but also on a better understanding of the underlying fraud mechanism. In addition, fraud being a criminal offence, the evidential aspect of the process must also be considered. These legal, operational, and strategic constraints lead to compromises on the means to be imple- mented for fraud management. This paper first focuses on the translation of practical questions raised in the banking industry at each step of the fraud management process into performance evaluation required to design a fraud detection model. Secondly, it considers a range of machine learning approaches that address these specificities: the imbalance between fraudulent and nonfraudulent operations, the lack of fully trusted labels, the concept-drift phenomenon, and the unavoidable trade-off between accuracy and interpretability of detection. -
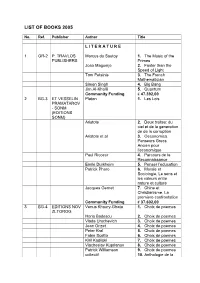
List of Books 2005
LIST OF BOOKS 2005 No. Ref. Publisher Author Title L I T E R A T U R E 1 GR-2 P. TRAVLOS Marcus du Sautoy 1. The Music of the PUBLISHERS Primes Joao Magueijo 2. Faster than the Speed of Light Tom Petsinis 3. The French Mathematician Simon Singh 4. Big Bang Jim Al-Khalili 5. Quantum Community Funding € 47.392,00 2 BG-3 ET VESSELIN Platon 1. Les Lois PRAMATAROV - SONM (EDITIONS SONM) Aristote 2. Deux traites: du ciel et de la generation de de la corruption Aristote et al 3. Oeconomica. Penseurs Grecs Ancien pour l’economique Paul Ricoeur 4. Parcours de la Reconnaissance Emile Durkheim 5. Penser l’education Patrick Pharo 6. Morale et Sociologie. Le sens et les valeurs entre nature et culture Jacques Gernet 7. Chine et Christianisme. La premiere confrontation Community Funding € 37.602,00 3 BG-4 EDITIONS NOV Venus Khoury-Ghata 1. Choix de poemes ZLTOROG Horia Badescu 2. Choix de poemes Vlada Urochevich 3. Choix de poemes Jean Orizet 4. Choix de poemes Peter Kral 5. Choix de poemes Fabio Scotto 6. Choix de poemes Kiril Kadiiski 7. Choix de poemes Viacheslav Kuprianov 8. Choix de poemes Patrick Williamson 9. Choix de poemes collectif 10. Anthologie de la poesie italienne du XX siecle (1950-2000) Community Funding € 48.000,00 4 N-15 H. Magda Szabo 1. Az Ajto ASCHEHOUG & CO Arto Paaslinna 2. Ulvova Myllaeri Susanna Clarke 3. Jonathon Strange & Mr. Norrell Helen Walsh 4. Brass Community Funding € 49.509,26 5 DK- TIDERNE Ernst Brunner 1. -

Identity Management Volume 103 No
Contents Telektronikk Identity Management Volume 103 No. 3/4 – 2007 1 Guest Editorial; ISSN 0085-7130 Do Van Thanh Editor: 3 The Ambiguity of Identity; Per Hjalmar Lehne Do Van Thanh, Ivar Jørstad (+47) 916 94 909 [email protected] 11 Identity Management Demystified; Do Van Thuan Editorial assistant: Gunhild Luke 19 Identity Management Standards, Systems and Standardisation Bodies; (+47) 415 14 125 Nguyen Duy Hinh, Sjur Millidahl, Do Van Thuan, Ivar Jørstad [email protected] 31 Identity Management in General and with Attention to Mobile GSM- Editorial office: based Systems; Tor Hjalmar Johannessen Telenor R&I NO-1331 Fornebu 52 Next Generation of Digital Identity; Norway Fulup Ar Foll, Jason Baragry (+47) 810 77 000 57 Microsoft Windows CardSpace and the Identity Metasystem; [email protected] Ole Tom Seierstad www.telektronikk.com 66 Smart Cards and Digital Identity; Editorial board: Jean-Daniel Aussel Berit Svendsen, Head of Telenor Nordic Fixed Ole P. Håkonsen, Professor NTNU 79 Trusting an eID in Open and International Communication; Oddvar Hesjedal, VP Telenor Mobile Operations Sverre Bauck Bjørn Løken, Director Telenor Nordic 85 Building a Federated Identity for Education: Feide; Graphic design: Ingrid Melve, Andreas Åkre Solberg Design Consult AS (Odd Andersen), Oslo 103 Identity Federation in a Multi Circle-of-Trust Constellation; Layout and illustrations: Dao Van Tran, Pål Løkstad, Do Van Thanh Gunhild Luke and Åse Aardal, 119 Identity Management in Telecommunication Business; Telenor R&I Do Van Thanh, -

Jean Sénac, Poet of the Algerian Revolution
City University of New York (CUNY) CUNY Academic Works All Dissertations, Theses, and Capstone Projects Dissertations, Theses, and Capstone Projects 10-2014 Jean Sénac, Poet of the Algerian Revolution Kai G. Krienke Graduate Center, City University of New York How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/gc_etds/438 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] Jean Sénac, Poet of the Algerian Revolution by Kai Krienke A dissertation submitted to the Graduate Faculty in Comparative Literature in partial fulfillment of the requirements for the degree of Doctor of Philosophy, The City University of New York. 2014 © 2014 KAI KRIENKE All Rights Reserved ii This manuscript has been read and accepted for the Graduate Faculty in Comparative Literature in satisfaction of the dissertation requirement for the degree of Doctor of Philosophy. Dr. Ammiel Alcalay 9/12/14 Date Chair of Examining Committee Dr. Giancarlo Lombardi 9/11/14 Date Executive Officer Dr. Ammiel Alcalay Dr. Andrea Khalil Dr. Caroline Rupprecht Supervisory Committee THE CITY UNIVERSITY OF NEW YORK iii Abstract Jean Sénac, poet of the Algerian Revolution by Kai Krienke Advisor: Ammiel Alcalay The work presented here is an exploration of the poetry and life of Jean Sénac, and through Sénac, of the larger role of poetry in the political and social movements of the 50s, 60s, and early 70s, mainly in Algeria and America. While Sénac was part of the European community in Algeria, his position regarding French rule changed dramatically over the course of the Algerian War, (between 1954 and 1962) and upon independence, he became one the rare French to return to his adopted homeland. -

From Quality Journalism to Speculative Journalism
04 transfer// 2 0 0 9 00 II Josep Lluís Gómez Mompart From quality journalism to speculative journalism Between January 2007 and May 2008, due to old age or sickness, among other reasons, quite a few journalists representative of what in the last century was considered quality journalism died: journalism that has not only honoured a profession necessary for the development and consolidation of democracy and freedoms, but which has also contributed, as the brilliant journalist Jean Daniel believes it should, to exposing everything that the authorities virtually always try to hide or, at least, divert attention from. Probably for this reason, this profession is “the best in the world”, in the words of Gabriel García Márquez, or “the most interesting”, after literature, according to Mario Vargas Llosa. I would like to remember half a dozen journalists, in some ways paradigmatic, of all those who passed away in those eighteen months. I shall begin with Ryszard Kapuscinski, the Polish reporter who died in Warsaw on January 23rd 2007, aged 75, an exemplary Face II (Rostre II), Jaume Plensa (2008). Mixed media and collage on paper, 220 x 200 cm 52/53 II From quality journalism to speculative journalism Josep Lluís Gómez Mompart communicator who has bequeathed an Kapuscinski was entire philosophy of being to a group at times rather sceptical and at others fascinated by journalism cynical: “in order to be a journalist you and by its possibilities have to be a good person” or “cynics are no use at this profession”, he would say of learning about the repeatedly.