MOB (Mps One Binder) Proteins in the Hippo Pathway and Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Detection of Aneuploidies by Paralogous Sequence Quantification S Deutsch, U Choudhury, G Merla, C Howald, a Sylvan, S E Antonarakis
908 J Med Genet: first published as 10.1136/jmg.2004.023184 on 9 December 2004. Downloaded from ORIGINAL ARTICLE Detection of aneuploidies by paralogous sequence quantification S Deutsch, U Choudhury, G Merla, C Howald, A Sylvan, S E Antonarakis ............................................................................................................................... J Med Genet 2004;41:908–915. doi: 10.1136/jmg.2004.023184 Background: Chromosomal aneuploidies are a common cause of congenital disorders associated with cognitive impairment and multiple dysmorphic features. Pre-natal diagnosis of aneuploidies is most See end of article for commonly performed by the karyotyping of fetal cells obtained by amniocentesis or chorionic villus authors’ affiliations sampling, but this method is labour intensive and requires about 14 days to complete. ....................... Methods: We have developed a PCR based method for the detection of targeted chromosome number Correspondence to: abnormalities termed paralogous sequence quantification (PSQ), based on the use of paralogous genes. Professor Stylianos E Paralogous sequences have a high degree of sequence identity, but accumulate nucleotide substitutions in Antonarakis, Department a locus specific manner. These sequence differences, which we term paralogous sequence mismatches of Genetic Medicine and Development, University of (PSMs), can be quantified using pyrosequencing technology, to estimate the relative dosage between Geneva Medical School, different chromosomes. We designed 10 assays for the detection of trisomies of chromosomes 13, 18, and GE 1211, Geneva, 21 and sex chromosome aneuploidies. Switzerland; Stylianos. antonarakis@medecine. Results: We evaluated the performance of this method on 175 DNAs, highly enriched for abnormal unige.ch samples. A correct and unambiguous diagnosis was given for 119 out of 120 aneuploid samples as well as for all the controls. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Novel Mutation and Three Other Sequence Variants Segregating with Phenotype at Keratoconus 13Q32 Susceptibility Locus
European Journal of Human Genetics (2012) 20, 389–397 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus Marta Czugala1,6, Justyna A Karolak1,6, Dorota M Nowak1, Piotr Polakowski2, Jose Pitarque3, Andrea Molinari3, Malgorzata Rydzanicz1, Bassem A Bejjani4, Beatrice YJT Yue5, Jacek P Szaflik2 and Marzena Gajecka*,1 Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A4C (p.Gln754His) and c.720+43A4GinDOCK9; c.2377-132A4CinIPO5 and c.1053+29G4CinSTK24. PolyPhen analyses predicted that c.2262A4C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A4C (p.Gln754His) -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

A Rhoa and Rnd3 Cycle Regulates Actin Reassembly During Membrane
A RhoA and Rnd3 cycle regulates actin reassembly PNAS PLUS during membrane blebbing Kana Aokia, Fumiyo Maedaa,1, Tomoya Nagasakob,1, Yuki Mochizukia, Seiichi Uchidab, and Junichi Ikenouchia,c,d,2 aDepartment of Biology, Faculty of Sciences, Kyushu University, Fukuoka 819-0395, Japan; bDepartment of Advanced Information Technology, Kyushu University, Fukuoka 819-0395, Japan; cPrecursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan; and dAMED-PRIME, Japan Agency for Medical Research and Development, Tokyo 100-0004, Japan Edited by Thomas D. Pollard, Yale University, New Haven, CT, and approved February 12, 2016 (received for review January 21, 2016) The actin cytoskeleton usually lies beneath the plasma membrane. membrane blebs is promoted by epidermal growth factor receptor When the membrane-associated actin cytoskeleton is transiently kinase substrate 8 (Eps8) and ezrin, and regulated by a RhoA– disrupted or the intracellular pressure is increased, the plasma Rho-associated protein kinase (ROCK)–Rnd3 feedback loop. membrane detaches from the cortex and protrudes. Such protruded membrane regions are called blebs. However, the molecular mech- Results and Discussion anisms underlying membrane blebbing are poorly understood. This Membrane Blebs Retract from Multiple Sites. We used the human study revealed that epidermal growth factor receptor kinase sub- colon carcinoma cell line DLD1 to observe membrane blebbing. strate 8 (Eps8) and ezrin are important regulators of rapid actin When cultured in 2D conditions, DLD1 cells did not exhibit reassembly for the initiation and retraction of protruded blebs. Live- membrane blebbing (Fig. 1A, Left). However, DLD1 cells ac- cell imaging of membrane blebbing revealed that local reassembly tively formed membrane blebs when embedded in a type I col- of actin filaments occurred at Eps8- and activated ezrin-positive foci lagen gel (Fig. -
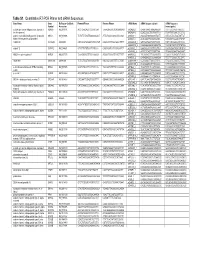
Table S1. Quantitative RT-PCR Primer and Sirna Sequences Gene Name
Table S1. Quantitative RT-PCR Primer and siRNA Sequences Gene Name Gene RefSeq or GenBank Forward Primer Reverse Primer siRNA Name siRNA Sequence (plus) siRNA Sequence Symbol Accession (minus/guide) a disintegrin and metalloproteinase domain 9 ADAM9 NM_003816 ACCTCAGCAGTTCCCATCAA TAAAGGAGGTGCAGGAGCAG siADAM9_1 GAGATTAACTAGAGAAAGA TCTTTCTCTAGTTAATCTC (meltrin gamma) siADAM9_2 GGAGGGAGTTCATAATTCA TGAATTATGAACTCCCTCC guanine nucleotide binding protein (G protein), GNAI3 NM_006496 TCTGTTATAGTTGGCGGCAGT ATTCTGCAAAGGCAAAAGGA siGNAI3_1 AGAACTAGCAGGAGTGATT AATCACTCCTGCTAGTTCT alpha inhibiting activity polypeptide 3 siGNAI3_2 ACACAGGTTCCAATACATA TATGTATTGGAACCTGTGT AA099748 AA099748 AA099748 CTACCACTGAGGTGTCCCTGT CAGCCATCTAACAGCATTTTT siAA099748_1 GTTAGATGGCTGATTAATA TATTAATCAGCCATCTAAC siAA099748_2 CGACAGAGGAGCAGCATTA TAATGCTGCTCCTCTGTCG calpain 12 CAPN12 NM_144691 GTCCTTCTGTCCCTCATCCA CAGCAGCTCCTCTGGAATCT siCAPN12_1 GGACACGCGTATTCCATCA TGATGGAATACGCGTGTCC siCAPN12_2 AATCCTCAGTTCCGTTTAA TTAAACGGAACTGAGGATT NMDA receptor regulated 1 NARG1 NM_057175 TAAAGGGAATTTGCCGAAGA AGCACTGGAGTTCGGTTTGT siNARG1_1 TGCGAGATCTTGAGGGTTA TAACCCTCAAGATCTCGCA siNARG1_2 GATAGGAGGTCCAAAAGAA TTCTTTTGGACCTCCTATC AK091308 AK091308 AK091308 TCTCCTGAATGGGAGGAATG GGCAACAGATGTTTCCTGTG siAK091308_1 CCAAAGACTTAGACTGTAA TTACAGTCTAAGTCTTTGG siAK091308_2 GCACAAAGCATTTACAACA TGTTGTAAATGCTTTGTGC serine/threonine kinase 24 (STE20 homolog, STK24 NM_003576 GCATCTGCCTTCCTTATCCA TGACAGTGTTTTGCCAGAGG siSTK24_1 TCGATTATCTCCATTCGGA TCCGAATGGAGATAATCGA yeast) siSTK24_2 CATCGGACTTGGACAGAAA -

Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia
Published OnlineFirst September 20, 2013; DOI: 10.1158/2159-8290.CD-13-0350 RESEARCH ARTICLE Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia Tea Pemovska 1 , Mika Kontro 2 , Bhagwan Yadav 1 , Henrik Edgren 1 , Samuli Eldfors1 , Agnieszka Szwajda 1 , Henrikki Almusa 1 , Maxim M. Bespalov 1 , Pekka Ellonen 1 , Erkki Elonen 2 , Bjørn T. Gjertsen5 , 6 , Riikka Karjalainen 1 , Evgeny Kulesskiy 1 , Sonja Lagström 1 , Anna Lehto 1 , Maija Lepistö1 , Tuija Lundán 3 , Muntasir Mamun Majumder 1 , Jesus M. Lopez Marti 1 , Pirkko Mattila 1 , Astrid Murumägi 1 , Satu Mustjoki 2 , Aino Palva 1 , Alun Parsons 1 , Tero Pirttinen 4 , Maria E. Rämet 4 , Minna Suvela 1 , Laura Turunen 1 , Imre Västrik 1 , Maija Wolf 1 , Jonathan Knowles 1 , Tero Aittokallio 1 , Caroline A. Heckman 1 , Kimmo Porkka 2 , Olli Kallioniemi 1 , and Krister Wennerberg 1 ABSTRACT We present an individualized systems medicine (ISM) approach to optimize cancer drug therapies one patient at a time. ISM is based on (i) molecular profi ling and ex vivo drug sensitivity and resistance testing (DSRT) of patients’ cancer cells to 187 oncology drugs, (ii) clinical implementation of therapies predicted to be effective, and (iii) studying consecutive samples from the treated patients to understand the basis of resistance. Here, application of ISM to 28 samples from patients with acute myeloid leukemia (AML) uncovered fi ve major taxonomic drug-response sub- types based on DSRT profi les, some with distinct genomic features (e.g., MLL gene fusions in subgroup IV and FLT3 -ITD mutations in subgroup V). Therapy based on DSRT resulted in several clinical responses. -

Analysis of Protein Complexes Through Model-Based Biclustering of Label-Free Quantitative AP-MS Data
Molecular Systems Biology 6; Article number 385; doi:10.1038/msb.2010.41 Citation: Molecular Systems Biology 6:385 & 2010 EMBO and Macmillan Publishers Limited All rights reserved 1744-4292/10 www.molecularsystemsbiology.com REPORT Analysis of protein complexes through model-based biclustering of label-free quantitative AP-MS data Hyungwon Choi1, Sinae Kim2, Anne-Claude Gingras3,4 and Alexey I Nesvizhskii1,5,* 1 Department of Pathology, University of Michigan, Ann Arbor, MI, USA, 2 Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA, 3 Samuel Lunenfeld Research Institute at Mount Sinai Hospital, Toronto, Ontario, Canada, 4 Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada and 5 Center for Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA * Corresponding author. Department of Pathology, University of Michigan, 1301 Catherine, 4237 MS1, Ann Arbor, MI 48109, USA. Tel.: þ 1 734 764 3516; Fax: þ 1 734 936 7361; E-mail: [email protected] Received 28.8.09; accepted 7.5.10 Affinity purification followed by mass spectrometry (AP-MS) has become a common approach for identifying protein–protein interactions (PPIs) and complexes. However, data analysis and visualization often rely on generic approaches that do not take advantage of the quantitative nature of AP-MS. We present a novel computational method, nested clustering, for biclustering of label-free quantitative AP-MS data. Our approach forms bait clusters based on the similarity of quantitative interaction profiles and identifies submatrices of prey proteins showing consistent quantitative association within bait clusters. In doing so, nested clustering effectively addresses the problem of overrepresentation of interactions involving baits proteins as compared with proteins only identified as preys. -

Large-Scale Rnai Screening Uncovers New Therapeutic Targets in the Human Parasite
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.05.935833; this version posted February 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Large-scale RNAi screening uncovers new therapeutic targets in the human parasite 2 Schistosoma mansoni 3 4 Jipeng Wang1*, Carlos Paz1*, Gilda Padalino2, Avril Coghlan3, Zhigang Lu3, Irina Gradinaru1, 5 Julie N.R. Collins1, Matthew Berriman3, Karl F. Hoffmann2, James J. Collins III1†. 6 7 8 1Department of Pharmacology, UT Southwestern Medical Center, Dallas, Texas 75390 9 2Institute of Biological, Environmental and Rural Sciences (IBERS), Aberystwyth University, 10 Aberystwyth, Wales, UK. 11 3Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SA, UK 12 *Equal Contribution 13 14 15 16 17 18 19 20 21 22 23 †To whom correspondence should be addressed 24 [email protected] 25 UT Southwestern Medical Center 26 Department of Pharmacology 27 6001 Forest Park. Rd. 28 Dallas, TX 75390 29 United States of America bioRxiv preprint doi: https://doi.org/10.1101/2020.02.05.935833; this version posted February 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 30 ABSTRACT 31 Schistosomes kill 250,000 people every year and are responsible for serious morbidity in 240 32 million of the world’s poorest people. -

Mirna‑26A‑5P and Mir‑26B‑5P Inhibit the Proliferation of Bladder Cancer Cells by Regulating PDCD10
ONCOLOGY REPORTS 40: 3523-3532, 2018 miRNA‑26a‑5p and miR‑26b‑5p inhibit the proliferation of bladder cancer cells by regulating PDCD10 KE WU1*, XING-YU MU1*, JUN-TAO JIANG1, MING-YUE TAN1, REN-JIE WANG1, WEN-JIE ZHOU2, XIANG WANG1, YIN-YAN HE3, MING-QING LI2 and ZHI-HONG LIU1 1Department of Urology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080; 2Laboratory for Reproductive Immunology, Hospital of Obstetrics and Gynecology, Fudan University, Shanghai 200011; 3Department of Obstetrics and Gynecology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, P.R. China Received October 11, 2017; Accepted September 4, 2018 DOI: 10.3892/or.2018.6734 Abstract. MicroRNA (miR)-26a-5p and miR-26b-5p consis- and miR-26b-5p were pivotal regulators in BC progression tently play an antitumor role in many types of cancers, but the by targeting the proliferation-related protein, PDCD10. The underlying mechanism remains unclear in bladder cancer (BC). miR-26-5p/PDCD10 interaction may provide important In the present study, we found that, in BC tissues, the levels of insight into the pathway of BC progression and present novel miR-26a-5p and miR-26b-5p were lower than in paired normal opportunities for future diagnosis and treatment strategies, tissues. The upregulation of miR‑26‑5p significantly inhibited especially for patients with high levels of PDCD10. the proliferation of BC cell lines (T24 and 5637). Bioinformatics analysis indicated that Programmed Cell Death 10 (PDCD10) Introduction was the downstream target gene of miR-26a-5p/miR-26b-5p, and this was ascertained by western blotting and quantitative Bladder cancer (BC) is one of the most important urinary real-time reverse transcription PCR (RT-qPCR).