Detection of Aneuploidies by Paralogous Sequence Quantification S Deutsch, U Choudhury, G Merla, C Howald, a Sylvan, S E Antonarakis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

NUFIP1 Sirna (H): Sc-105367
SANTA CRUZ BIOTECHNOLOGY, INC. NUFIP1 siRNA (h): sc-105367 BACKGROUND STORAGE AND RESUSPENSION NUFIP1 (nuclear fragile X mental retardation-interacting protein 1) is a 495 Store lyophilized siRNA duplex at -20° C with desiccant. Stable for at least amino acid protein that localizes to the nucleus and can interact with FMR1 one year from the date of shipment. Once resuspended, store at -20° C, (fragile X mental retardation protein) and BRCA1, a breast and ovarian-specific avoid contact with RNAses and repeated freeze thaw cycles. tumor suppressor. Through its interaction with FMR1, NUFIP1 is thought to Resuspend lyophilized siRNA duplex in 330 µl of the RNAse-free water shuttle specific mRNPs to active neuronal synapses, thereby regulating the provided. Resuspension of the siRNA duplex in 330 µl of RNAse-free water translation of synaptic plasticity-related mRNA. The close interaction of makes a 10 µM solution in a 10 µM Tris-HCl, pH 8.0, 20 mM NaCl, 1 mM NUFIP1 with FMR1, a protein that is essential for proper dendritic spine matu- EDTA buffered solution. ration, suggests close involvement in neuronal development. Interaction of NUFIP1 with BRCA1 results in the formation of a complex which binds the APPLICATIONS positive elongation factor P-TEFb, thus stimulating RNA polymerase II (pol II) transcription. When associated with BRAC1, NUFIP1 acts as a transcriptional NUFIP1 siRNA (h) is recommended for the inhibition of NUFIP1 expression in activator contributing to tumor suppressor gene expression. NUFIP1 contains human cells. one C2H2-type zinc finger and is expressed throughout the body. SUPPORT REAGENTS REFERENCES For optimal siRNA transfection efficiency, Santa Cruz Biotechnology’s 1. -

Novel Mutation and Three Other Sequence Variants Segregating with Phenotype at Keratoconus 13Q32 Susceptibility Locus
European Journal of Human Genetics (2012) 20, 389–397 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus Marta Czugala1,6, Justyna A Karolak1,6, Dorota M Nowak1, Piotr Polakowski2, Jose Pitarque3, Andrea Molinari3, Malgorzata Rydzanicz1, Bassem A Bejjani4, Beatrice YJT Yue5, Jacek P Szaflik2 and Marzena Gajecka*,1 Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A4C (p.Gln754His) and c.720+43A4GinDOCK9; c.2377-132A4CinIPO5 and c.1053+29G4CinSTK24. PolyPhen analyses predicted that c.2262A4C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A4C (p.Gln754His) -
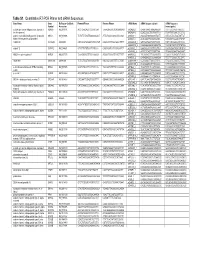
Table S1. Quantitative RT-PCR Primer and Sirna Sequences Gene Name
Table S1. Quantitative RT-PCR Primer and siRNA Sequences Gene Name Gene RefSeq or GenBank Forward Primer Reverse Primer siRNA Name siRNA Sequence (plus) siRNA Sequence Symbol Accession (minus/guide) a disintegrin and metalloproteinase domain 9 ADAM9 NM_003816 ACCTCAGCAGTTCCCATCAA TAAAGGAGGTGCAGGAGCAG siADAM9_1 GAGATTAACTAGAGAAAGA TCTTTCTCTAGTTAATCTC (meltrin gamma) siADAM9_2 GGAGGGAGTTCATAATTCA TGAATTATGAACTCCCTCC guanine nucleotide binding protein (G protein), GNAI3 NM_006496 TCTGTTATAGTTGGCGGCAGT ATTCTGCAAAGGCAAAAGGA siGNAI3_1 AGAACTAGCAGGAGTGATT AATCACTCCTGCTAGTTCT alpha inhibiting activity polypeptide 3 siGNAI3_2 ACACAGGTTCCAATACATA TATGTATTGGAACCTGTGT AA099748 AA099748 AA099748 CTACCACTGAGGTGTCCCTGT CAGCCATCTAACAGCATTTTT siAA099748_1 GTTAGATGGCTGATTAATA TATTAATCAGCCATCTAAC siAA099748_2 CGACAGAGGAGCAGCATTA TAATGCTGCTCCTCTGTCG calpain 12 CAPN12 NM_144691 GTCCTTCTGTCCCTCATCCA CAGCAGCTCCTCTGGAATCT siCAPN12_1 GGACACGCGTATTCCATCA TGATGGAATACGCGTGTCC siCAPN12_2 AATCCTCAGTTCCGTTTAA TTAAACGGAACTGAGGATT NMDA receptor regulated 1 NARG1 NM_057175 TAAAGGGAATTTGCCGAAGA AGCACTGGAGTTCGGTTTGT siNARG1_1 TGCGAGATCTTGAGGGTTA TAACCCTCAAGATCTCGCA siNARG1_2 GATAGGAGGTCCAAAAGAA TTCTTTTGGACCTCCTATC AK091308 AK091308 AK091308 TCTCCTGAATGGGAGGAATG GGCAACAGATGTTTCCTGTG siAK091308_1 CCAAAGACTTAGACTGTAA TTACAGTCTAAGTCTTTGG siAK091308_2 GCACAAAGCATTTACAACA TGTTGTAAATGCTTTGTGC serine/threonine kinase 24 (STE20 homolog, STK24 NM_003576 GCATCTGCCTTCCTTATCCA TGACAGTGTTTTGCCAGAGG siSTK24_1 TCGATTATCTCCATTCGGA TCCGAATGGAGATAATCGA yeast) siSTK24_2 CATCGGACTTGGACAGAAA -

Identification and Characterization of Novel Fusion Genes with Potential
International Journal of Molecular Sciences Article Identification and Characterization of Novel Fusion Genes with Potential Clinical Applications in Mexican Children with Acute Lymphoblastic Leukemia Minerva Mata-Rocha 1,2 , Angelica Rangel-López 3 , Elva Jiménez-Hernández 4,5, Blanca Angélica Morales-Castillo 6, Carolina González-Torres 7, Javier Gaytan-Cervantes 7, Enrique Álvarez-Olmos 6, Juan Carlos Núñez-Enríquez 6 , Arturo Fajardo-Gutiérrez 6, Jorge Alfonso Martín-Trejo 8, Karina Anastacia Solís-Labastida 8, Aurora Medina-Sansón 9, Janet Flores-Lujano 6, Omar Alejandro Sepúlveda-Robles 1,2 , José Gabriel Peñaloza-González 10, Laura Eugenia Espinoza-Hernández 4, Nora Nancy Núñez-Villegas 4, Rosa Martha Espinosa-Elizondo 11, Beatriz Cortés-Herrera 11, José Refugio Torres-Nava 5, Luz Victoria Flores-Villegas 12, Laura Elizabeth Merino-Pasaye 12, Vilma Carolina Bekker-Méndez 13, Martha Margarita Velázquez-Aviña 10, María Luisa Pérez-Saldívar 6, Benito Alejandro Bautista-Martínez 8, Raquel Amador-Sánchez 14, Ana Itamar González-Avila 14, Silvia Jiménez-Morales 15 , David Aldebarán Duarte-Rodríguez 6, Jessica Denisse Santillán-Juárez 16, Alejandra Jimena García-Velázquez 16, Haydeé Rosas-Vargas 2,* and Juan Manuel Mejía-Aranguré 17,* 1 CONACyT-Unidad de Investigacion Medica en Epidemiologia Clinica, Hospital de Pediatria, Centro Medico Siglo XXI, IMSS, 06720 Mexico City, Mexico; [email protected] (M.M.-R.); [email protected] (O.A.S.-R.) 2 Unidad de Investigacion Medica en Genética Humana, Hospital de Pediatria, Centro Medico Nacional -

Integrative Genomics Analyses Reveal Molecularly Distinct Subgroups of B-Cell Chronic Lymphocytic Leukemia Patients with 13Q14 Deletion
Published OnlineFirst October 14, 2010; DOI: 10.1158/1078-0432.CCR-10-0151 Clinical Cancer Human Cancer Biology Research Integrative Genomics Analyses Reveal Molecularly Distinct Subgroups of B-Cell Chronic Lymphocytic Leukemia Patients with 13q14 Deletion Laura Mosca1, Sonia Fabris1, Marta Lionetti1, Katia Todoerti1, Luca Agnelli1, Fortunato Morabito2, Giovanna Cutrona3, Adrian Andronache1, Serena Matis3, Francesco Ferrari4, Massimo Gentile2, Mauro Spriano5, Vincenzo Callea6, Gianluca Festini7, Stefano Molica8, Giorgio Lambertenghi Deliliers1, Silvio Bicciato4, Manlio Ferrarini3,9, and Antonino Neri1 Abstract Purpose: Chromosome 13q14 deletion occurs in a substantial number of chronic lymphocytic leukemia (CLL) patients and it is believed to play a pathogenetic role. The exact mechanisms involved in this lesion have not yet been fully elucidated because of its heterogeneity and the imprecise knowledge of the implicated genes. This study was addressed to further contribute to the molecular definition of this lesion in CLL. Experimental Design: We applied single-nucleotide polymorphism (SNP)-array technology and gene expression profiling data to investigate the 13q14 deletion occurring in a panel of 100 untreated, early-stage (Binet A) patients representative of the major genetics, molecular, and biological features of the disease. Results: Concordantly with FISH analysis, SNP arrays identified 44 patients with del(13)(q14) including 11 cases with a biallelic deletion. The shorter monoallelic deletion was 635-kb long. The loss of the miR-15a/16-1 cluster occurred in all del(13)(q14) cases except in 2 patients with a monoallelic deletion, who retained both copies. MiR-15a/16 expression was significantly downregulated only in patients with the biallelic loss of the miRNA cluster compared to 13q normal cases. -

STK24 Antibody - C-Terminal Region Rabbit Polyclonal Antibody Catalog # AI16132
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 STK24 Antibody - C-terminal region Rabbit Polyclonal Antibody Catalog # AI16132 Specification STK24 Antibody - C-terminal region - Product Information Application WB Primary Accession Q9Y6E0 Other Accession NP_003567 Reactivity Human Host Rabbit Clonality Polyclonal Calculated MW 48kDa KDa STK24 Antibody - C-terminal region - Additional Information Host: Rabbit Target Name: STK24 Gene ID 8428 Sample Tissue: 293T Whole Cell lysates Antibody Dilution: 1.0μg/ml Alias Symbol STK24, MST3, STK3, Other Names STK24 Antibody - C-terminal region - Serine/threonine-protein kinase 24, Background 2.7.11.1, Mammalian STE20-like protein kinase 3, MST-3, STE20-like kinase MST3, Serine/threonine-protein kinase that acts on Serine/threonine-protein kinase 24 36 kDa both serine and threonine residues and subunit, Mammalian STE20-like protein promotes apoptosis in response to stress kinase 3 N-terminal, MST3/N, stimuli and caspase activation. Mediates Serine/threonine-protein kinase 24 12 kDa oxidative-stress- induced cell death by subunit, Mammalian STE20-like protein modulating phosphorylation of JNK1-JNK2 kinase 3 C-terminal, MST3/C, STK24, MST3, (MAPK8 and MAPK9), p38 (MAPK11, MAPK12, STK3 MAPK13 and MAPK14) during oxidative stress. Plays a role in a staurosporine-induced Format caspase- independent apoptotic pathway by Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and regulating the nuclear translocation of AIFM1 2% sucrose. and ENDOG and the DNase activity associated with ENDOG. Phosphorylates STK38L on Reconstitution & Storage 'Thr-442' and stimulates its kinase activity. Add 50 &mu, l of distilled water. -

MST3 (STK24) (NM 001032296) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC209117L4V MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Symbol: STK24 Synonyms: HEL-S-95; MST3; MST3B; STE20; STK3 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_001032296 ORF Size: 1293 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC209117). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_001032296.2 RefSeq Size: 4624 bp RefSeq ORF: 1296 bp Locus ID: 8428 UniProt ID: Q9Y6E0, Q5U0E6, Q6P0Y1 Protein Families: Druggable Genome, Protein Kinase MW: 47.7 kDa Gene Summary: This gene encodes a serine/threonine protein kinase that functions upstream of mitogen- activated protein kinase (MAPK) signaling. The encoded protein is cleaved into two chains by caspases; the N-terminal fragment (MST3/N) translocates to the nucleus and promotes programmed cells death. -

STRIPAK Complexes in Cell Signaling and Cancer
Oncogene (2016), 1–9 © 2016 Macmillan Publishers Limited All rights reserved 0950-9232/16 www.nature.com/onc REVIEW STRIPAK complexes in cell signaling and cancer Z Shi1,2, S Jiao1 and Z Zhou1,3 Striatin-interacting phosphatase and kinase (STRIPAK) complexes are striatin-centered multicomponent supramolecular structures containing both kinases and phosphatases. STRIPAK complexes are evolutionarily conserved and have critical roles in protein (de) phosphorylation. Recent studies indicate that STRIPAK complexes are emerging mediators and regulators of multiple vital signaling pathways including Hippo, MAPK (mitogen-activated protein kinase), nuclear receptor and cytoskeleton remodeling. Different types of STRIPAK complexes are extensively involved in a variety of fundamental biological processes ranging from cell growth, differentiation, proliferation and apoptosis to metabolism, immune regulation and tumorigenesis. Growing evidence correlates dysregulation of STRIPAK complexes with human diseases including cancer. In this review, we summarize the current understanding of the assembly and functions of STRIPAK complexes, with a special focus on cell signaling and cancer. Oncogene advance online publication, 15 February 2016; doi:10.1038/onc.2016.9 INTRODUCTION in the central nervous system and STRN4 is mostly abundant in Recent proteomic studies identified a group of novel multi- the brain and lung, whereas STRN3 is ubiquitously expressed in 5–9 component complexes named striatin (STRN)-interacting phos- almost all tissues. STRNs share a -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

STRIPAK Directs PP2A Activity Toward MAP4K4 to Promote Oncogenic Transformation
bioRxiv preprint doi: https://doi.org/10.1101/823096; this version posted October 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation Jong Wook Kim1,2,7,8*, Christian Berrios2,4*, Miju Kim1,2*, Amy E. Schade2,4*, Guillaume Adelmant5, Huwate Yeerna7, Emily Damato1, Amanda Balboni Iniguez1,6, Selene K. Swanson9, Laurence Florens9, Michael P. Washburn9.10, Kim Stegmaier1,6, Nathaniel S. Gray5, Pablo Tamayo7,8, Ole Gjoerup2, Jarrod A. Marto5, James A. DeCaprio2,3,4,†, William C. Hahn1, 2, 3,† 1Broad Institute of Harvard and MIT, Cambridge MA. 2Department of Medical Oncology, Dana-Farber Cancer Institute, Boston MA. 3Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston MA. 4Program in Virology, Graduate School of Arts and Sciences, Harvard University, Cambridge, MA. 5Department of Cancer Biology and Blais Proteomics Center, Dana-Farber Cancer Institute, Boston MA. 6Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston MA. 7Division of Medical Genetics, School of Medicine, UC San Diego, CA. 8Moores Cancer Center, University of California, San Diego, CA. 9Stowers Institute for Medical Research, Kansas City, MO 64110 10Department of Pathology and Laboratory Medicine, University of Kansas Medical Center, Kansas City, KS 66160 * These authors contributed equally to the work. †Co-senior authors 1 bioRxiv preprint doi: https://doi.org/10.1101/823096; this version posted October 29, 2019. -

Genotype-Phenotype Correlations in Patients with Retinoblastoma And
Genotype-phenotype correlations in patients with Retinoblastoma and an interstitial 13q deletion Diana Mitter, Reinhard Ullmann, Artur Muradyan, Ludger Klein-Hitpaß, Deniz Kanber, Dietmar R Lohmann, Katrin Ounap, Marc Kaulisch To cite this version: Diana Mitter, Reinhard Ullmann, Artur Muradyan, Ludger Klein-Hitpaß, Deniz Kanber, et al.. Genotype-phenotype correlations in patients with Retinoblastoma and an interstitial 13q deletion. European Journal of Human Genetics, Nature Publishing Group, 2011, 10.1038/ejhg.2011.58. hal- 00633984 HAL Id: hal-00633984 https://hal.archives-ouvertes.fr/hal-00633984 Submitted on 20 Oct 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Genotype-phenotype correlations in patients with Retinoblastoma and interstitial 13q deletions 1, *Diana Mitter, 2Reinhard Ullmann, 2Artur Muradyan, 3Ludger Klein-Hitpaß, 1Deniz Kanber, 4Katrin Õunap, 5Marc Kaulisch, 1Dietmar Lohmann 1Institut für Humangenetik, Universitätsklinikum Essen, Germany; 2Max-Planck- Institute of Molecular Genetic, Berlin, Germany; 3Institute for Cell Biology (Tumor Research), University of Duisburg-Essen, Germany; 4Department of Genetics, United Laboratories, Tartu University Hospital, Estonia; 5Institute for Research Information and Quality Assurance, Germany. *Correspondence and present address: Diana Mitter, Institut für Humangenetik, Universitätsklinikum Leipzig, Philipp-Rosenthal-Str.