Cerebral Cavernous Malformation Proteins in Barrier Maintenance and Regulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Detection of Aneuploidies by Paralogous Sequence Quantification S Deutsch, U Choudhury, G Merla, C Howald, a Sylvan, S E Antonarakis
908 J Med Genet: first published as 10.1136/jmg.2004.023184 on 9 December 2004. Downloaded from ORIGINAL ARTICLE Detection of aneuploidies by paralogous sequence quantification S Deutsch, U Choudhury, G Merla, C Howald, A Sylvan, S E Antonarakis ............................................................................................................................... J Med Genet 2004;41:908–915. doi: 10.1136/jmg.2004.023184 Background: Chromosomal aneuploidies are a common cause of congenital disorders associated with cognitive impairment and multiple dysmorphic features. Pre-natal diagnosis of aneuploidies is most See end of article for commonly performed by the karyotyping of fetal cells obtained by amniocentesis or chorionic villus authors’ affiliations sampling, but this method is labour intensive and requires about 14 days to complete. ....................... Methods: We have developed a PCR based method for the detection of targeted chromosome number Correspondence to: abnormalities termed paralogous sequence quantification (PSQ), based on the use of paralogous genes. Professor Stylianos E Paralogous sequences have a high degree of sequence identity, but accumulate nucleotide substitutions in Antonarakis, Department a locus specific manner. These sequence differences, which we term paralogous sequence mismatches of Genetic Medicine and Development, University of (PSMs), can be quantified using pyrosequencing technology, to estimate the relative dosage between Geneva Medical School, different chromosomes. We designed 10 assays for the detection of trisomies of chromosomes 13, 18, and GE 1211, Geneva, 21 and sex chromosome aneuploidies. Switzerland; Stylianos. antonarakis@medecine. Results: We evaluated the performance of this method on 175 DNAs, highly enriched for abnormal unige.ch samples. A correct and unambiguous diagnosis was given for 119 out of 120 aneuploid samples as well as for all the controls. -

Novel Mutation and Three Other Sequence Variants Segregating with Phenotype at Keratoconus 13Q32 Susceptibility Locus
European Journal of Human Genetics (2012) 20, 389–397 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus Marta Czugala1,6, Justyna A Karolak1,6, Dorota M Nowak1, Piotr Polakowski2, Jose Pitarque3, Andrea Molinari3, Malgorzata Rydzanicz1, Bassem A Bejjani4, Beatrice YJT Yue5, Jacek P Szaflik2 and Marzena Gajecka*,1 Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A4C (p.Gln754His) and c.720+43A4GinDOCK9; c.2377-132A4CinIPO5 and c.1053+29G4CinSTK24. PolyPhen analyses predicted that c.2262A4C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A4C (p.Gln754His) -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

A Rhoa and Rnd3 Cycle Regulates Actin Reassembly During Membrane
A RhoA and Rnd3 cycle regulates actin reassembly PNAS PLUS during membrane blebbing Kana Aokia, Fumiyo Maedaa,1, Tomoya Nagasakob,1, Yuki Mochizukia, Seiichi Uchidab, and Junichi Ikenouchia,c,d,2 aDepartment of Biology, Faculty of Sciences, Kyushu University, Fukuoka 819-0395, Japan; bDepartment of Advanced Information Technology, Kyushu University, Fukuoka 819-0395, Japan; cPrecursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan; and dAMED-PRIME, Japan Agency for Medical Research and Development, Tokyo 100-0004, Japan Edited by Thomas D. Pollard, Yale University, New Haven, CT, and approved February 12, 2016 (received for review January 21, 2016) The actin cytoskeleton usually lies beneath the plasma membrane. membrane blebs is promoted by epidermal growth factor receptor When the membrane-associated actin cytoskeleton is transiently kinase substrate 8 (Eps8) and ezrin, and regulated by a RhoA– disrupted or the intracellular pressure is increased, the plasma Rho-associated protein kinase (ROCK)–Rnd3 feedback loop. membrane detaches from the cortex and protrudes. Such protruded membrane regions are called blebs. However, the molecular mech- Results and Discussion anisms underlying membrane blebbing are poorly understood. This Membrane Blebs Retract from Multiple Sites. We used the human study revealed that epidermal growth factor receptor kinase sub- colon carcinoma cell line DLD1 to observe membrane blebbing. strate 8 (Eps8) and ezrin are important regulators of rapid actin When cultured in 2D conditions, DLD1 cells did not exhibit reassembly for the initiation and retraction of protruded blebs. Live- membrane blebbing (Fig. 1A, Left). However, DLD1 cells ac- cell imaging of membrane blebbing revealed that local reassembly tively formed membrane blebs when embedded in a type I col- of actin filaments occurred at Eps8- and activated ezrin-positive foci lagen gel (Fig. -
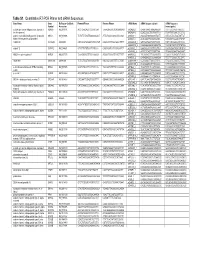
Table S1. Quantitative RT-PCR Primer and Sirna Sequences Gene Name
Table S1. Quantitative RT-PCR Primer and siRNA Sequences Gene Name Gene RefSeq or GenBank Forward Primer Reverse Primer siRNA Name siRNA Sequence (plus) siRNA Sequence Symbol Accession (minus/guide) a disintegrin and metalloproteinase domain 9 ADAM9 NM_003816 ACCTCAGCAGTTCCCATCAA TAAAGGAGGTGCAGGAGCAG siADAM9_1 GAGATTAACTAGAGAAAGA TCTTTCTCTAGTTAATCTC (meltrin gamma) siADAM9_2 GGAGGGAGTTCATAATTCA TGAATTATGAACTCCCTCC guanine nucleotide binding protein (G protein), GNAI3 NM_006496 TCTGTTATAGTTGGCGGCAGT ATTCTGCAAAGGCAAAAGGA siGNAI3_1 AGAACTAGCAGGAGTGATT AATCACTCCTGCTAGTTCT alpha inhibiting activity polypeptide 3 siGNAI3_2 ACACAGGTTCCAATACATA TATGTATTGGAACCTGTGT AA099748 AA099748 AA099748 CTACCACTGAGGTGTCCCTGT CAGCCATCTAACAGCATTTTT siAA099748_1 GTTAGATGGCTGATTAATA TATTAATCAGCCATCTAAC siAA099748_2 CGACAGAGGAGCAGCATTA TAATGCTGCTCCTCTGTCG calpain 12 CAPN12 NM_144691 GTCCTTCTGTCCCTCATCCA CAGCAGCTCCTCTGGAATCT siCAPN12_1 GGACACGCGTATTCCATCA TGATGGAATACGCGTGTCC siCAPN12_2 AATCCTCAGTTCCGTTTAA TTAAACGGAACTGAGGATT NMDA receptor regulated 1 NARG1 NM_057175 TAAAGGGAATTTGCCGAAGA AGCACTGGAGTTCGGTTTGT siNARG1_1 TGCGAGATCTTGAGGGTTA TAACCCTCAAGATCTCGCA siNARG1_2 GATAGGAGGTCCAAAAGAA TTCTTTTGGACCTCCTATC AK091308 AK091308 AK091308 TCTCCTGAATGGGAGGAATG GGCAACAGATGTTTCCTGTG siAK091308_1 CCAAAGACTTAGACTGTAA TTACAGTCTAAGTCTTTGG siAK091308_2 GCACAAAGCATTTACAACA TGTTGTAAATGCTTTGTGC serine/threonine kinase 24 (STE20 homolog, STK24 NM_003576 GCATCTGCCTTCCTTATCCA TGACAGTGTTTTGCCAGAGG siSTK24_1 TCGATTATCTCCATTCGGA TCCGAATGGAGATAATCGA yeast) siSTK24_2 CATCGGACTTGGACAGAAA -

Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia
Published OnlineFirst September 20, 2013; DOI: 10.1158/2159-8290.CD-13-0350 RESEARCH ARTICLE Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia Tea Pemovska 1 , Mika Kontro 2 , Bhagwan Yadav 1 , Henrik Edgren 1 , Samuli Eldfors1 , Agnieszka Szwajda 1 , Henrikki Almusa 1 , Maxim M. Bespalov 1 , Pekka Ellonen 1 , Erkki Elonen 2 , Bjørn T. Gjertsen5 , 6 , Riikka Karjalainen 1 , Evgeny Kulesskiy 1 , Sonja Lagström 1 , Anna Lehto 1 , Maija Lepistö1 , Tuija Lundán 3 , Muntasir Mamun Majumder 1 , Jesus M. Lopez Marti 1 , Pirkko Mattila 1 , Astrid Murumägi 1 , Satu Mustjoki 2 , Aino Palva 1 , Alun Parsons 1 , Tero Pirttinen 4 , Maria E. Rämet 4 , Minna Suvela 1 , Laura Turunen 1 , Imre Västrik 1 , Maija Wolf 1 , Jonathan Knowles 1 , Tero Aittokallio 1 , Caroline A. Heckman 1 , Kimmo Porkka 2 , Olli Kallioniemi 1 , and Krister Wennerberg 1 ABSTRACT We present an individualized systems medicine (ISM) approach to optimize cancer drug therapies one patient at a time. ISM is based on (i) molecular profi ling and ex vivo drug sensitivity and resistance testing (DSRT) of patients’ cancer cells to 187 oncology drugs, (ii) clinical implementation of therapies predicted to be effective, and (iii) studying consecutive samples from the treated patients to understand the basis of resistance. Here, application of ISM to 28 samples from patients with acute myeloid leukemia (AML) uncovered fi ve major taxonomic drug-response sub- types based on DSRT profi les, some with distinct genomic features (e.g., MLL gene fusions in subgroup IV and FLT3 -ITD mutations in subgroup V). Therapy based on DSRT resulted in several clinical responses. -

Analysis of Protein Complexes Through Model-Based Biclustering of Label-Free Quantitative AP-MS Data
Molecular Systems Biology 6; Article number 385; doi:10.1038/msb.2010.41 Citation: Molecular Systems Biology 6:385 & 2010 EMBO and Macmillan Publishers Limited All rights reserved 1744-4292/10 www.molecularsystemsbiology.com REPORT Analysis of protein complexes through model-based biclustering of label-free quantitative AP-MS data Hyungwon Choi1, Sinae Kim2, Anne-Claude Gingras3,4 and Alexey I Nesvizhskii1,5,* 1 Department of Pathology, University of Michigan, Ann Arbor, MI, USA, 2 Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA, 3 Samuel Lunenfeld Research Institute at Mount Sinai Hospital, Toronto, Ontario, Canada, 4 Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada and 5 Center for Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA * Corresponding author. Department of Pathology, University of Michigan, 1301 Catherine, 4237 MS1, Ann Arbor, MI 48109, USA. Tel.: þ 1 734 764 3516; Fax: þ 1 734 936 7361; E-mail: [email protected] Received 28.8.09; accepted 7.5.10 Affinity purification followed by mass spectrometry (AP-MS) has become a common approach for identifying protein–protein interactions (PPIs) and complexes. However, data analysis and visualization often rely on generic approaches that do not take advantage of the quantitative nature of AP-MS. We present a novel computational method, nested clustering, for biclustering of label-free quantitative AP-MS data. Our approach forms bait clusters based on the similarity of quantitative interaction profiles and identifies submatrices of prey proteins showing consistent quantitative association within bait clusters. In doing so, nested clustering effectively addresses the problem of overrepresentation of interactions involving baits proteins as compared with proteins only identified as preys. -

Mirna‑26A‑5P and Mir‑26B‑5P Inhibit the Proliferation of Bladder Cancer Cells by Regulating PDCD10
ONCOLOGY REPORTS 40: 3523-3532, 2018 miRNA‑26a‑5p and miR‑26b‑5p inhibit the proliferation of bladder cancer cells by regulating PDCD10 KE WU1*, XING-YU MU1*, JUN-TAO JIANG1, MING-YUE TAN1, REN-JIE WANG1, WEN-JIE ZHOU2, XIANG WANG1, YIN-YAN HE3, MING-QING LI2 and ZHI-HONG LIU1 1Department of Urology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080; 2Laboratory for Reproductive Immunology, Hospital of Obstetrics and Gynecology, Fudan University, Shanghai 200011; 3Department of Obstetrics and Gynecology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, P.R. China Received October 11, 2017; Accepted September 4, 2018 DOI: 10.3892/or.2018.6734 Abstract. MicroRNA (miR)-26a-5p and miR-26b-5p consis- and miR-26b-5p were pivotal regulators in BC progression tently play an antitumor role in many types of cancers, but the by targeting the proliferation-related protein, PDCD10. The underlying mechanism remains unclear in bladder cancer (BC). miR-26-5p/PDCD10 interaction may provide important In the present study, we found that, in BC tissues, the levels of insight into the pathway of BC progression and present novel miR-26a-5p and miR-26b-5p were lower than in paired normal opportunities for future diagnosis and treatment strategies, tissues. The upregulation of miR‑26‑5p significantly inhibited especially for patients with high levels of PDCD10. the proliferation of BC cell lines (T24 and 5637). Bioinformatics analysis indicated that Programmed Cell Death 10 (PDCD10) Introduction was the downstream target gene of miR-26a-5p/miR-26b-5p, and this was ascertained by western blotting and quantitative Bladder cancer (BC) is one of the most important urinary real-time reverse transcription PCR (RT-qPCR). -

STRIP1, a Core Component of STRIPAK Complexes, Is Essential
STRIP1, a core component of STRIPAK complexes, is PNAS PLUS essential for normal mesoderm migration in the mouse embryo Hisham Bazzia,b,c,1, Ekaterina Sorokab,c, Heather L. Alcorna, and Kathryn V. Andersona,1 aDevelopmental Biology Program, Sloan Kettering Institute, New York, NY 10065; bDepartment of Dermatology and Venereology, University Hospital of Cologne, 50937 Cologne, Germany; and cCologne Cluster of Excellence in Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, 50931 Cologne, Germany Edited by Brigid L. M. Hogan, Duke University Medical Center, Durham, NC, and approved November 10, 2017 (received for review August 1, 2017) Regulated mesoderm migration is necessary for the proper mor- E-cadherin is essential for mesoderm migration (9, 10). In the phogenesis and organ formation during embryonic development. chick, directional migration of the nascent mesoderm appears Cell migration and its dependence on the cytoskeleton and signaling to depend on chemorepulsion by FGF and Wnt3a ligands expressed machines have been studied extensively in cultured cells; in at the primitive streak (11), but it is not clear whether the same contrast, remarkably little is known about the mechanisms that signals operate in the mouse. The ability of mesoderm cells to mi- regulate mesoderm cell migration in vivo. Here, we report the grate depends on a complete reorganization of the actin cytoskel- identification and characterization of a mouse mutation in striatin- eton, and motility depends on the WAVE complex and the small Strip1 interacting protein 1 ( ) that disrupts migration of the meso- GTPase RAC1 (12, 13). derm after the gastrulation epithelial-to-mesenchymal transition Experiments in cell culture recently implicated the striatin- (EMT). -

STK24 Antibody - C-Terminal Region Rabbit Polyclonal Antibody Catalog # AI16132
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 STK24 Antibody - C-terminal region Rabbit Polyclonal Antibody Catalog # AI16132 Specification STK24 Antibody - C-terminal region - Product Information Application WB Primary Accession Q9Y6E0 Other Accession NP_003567 Reactivity Human Host Rabbit Clonality Polyclonal Calculated MW 48kDa KDa STK24 Antibody - C-terminal region - Additional Information Host: Rabbit Target Name: STK24 Gene ID 8428 Sample Tissue: 293T Whole Cell lysates Antibody Dilution: 1.0μg/ml Alias Symbol STK24, MST3, STK3, Other Names STK24 Antibody - C-terminal region - Serine/threonine-protein kinase 24, Background 2.7.11.1, Mammalian STE20-like protein kinase 3, MST-3, STE20-like kinase MST3, Serine/threonine-protein kinase that acts on Serine/threonine-protein kinase 24 36 kDa both serine and threonine residues and subunit, Mammalian STE20-like protein promotes apoptosis in response to stress kinase 3 N-terminal, MST3/N, stimuli and caspase activation. Mediates Serine/threonine-protein kinase 24 12 kDa oxidative-stress- induced cell death by subunit, Mammalian STE20-like protein modulating phosphorylation of JNK1-JNK2 kinase 3 C-terminal, MST3/C, STK24, MST3, (MAPK8 and MAPK9), p38 (MAPK11, MAPK12, STK3 MAPK13 and MAPK14) during oxidative stress. Plays a role in a staurosporine-induced Format caspase- independent apoptotic pathway by Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and regulating the nuclear translocation of AIFM1 2% sucrose. and ENDOG and the DNase activity associated with ENDOG. Phosphorylates STK38L on Reconstitution & Storage 'Thr-442' and stimulates its kinase activity. Add 50 &mu, l of distilled water. -

MST3 (STK24) (NM 001032296) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC209117L4V MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Symbol: STK24 Synonyms: HEL-S-95; MST3; MST3B; STE20; STK3 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_001032296 ORF Size: 1293 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC209117). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_001032296.2 RefSeq Size: 4624 bp RefSeq ORF: 1296 bp Locus ID: 8428 UniProt ID: Q9Y6E0, Q5U0E6, Q6P0Y1 Protein Families: Druggable Genome, Protein Kinase MW: 47.7 kDa Gene Summary: This gene encodes a serine/threonine protein kinase that functions upstream of mitogen- activated protein kinase (MAPK) signaling. The encoded protein is cleaved into two chains by caspases; the N-terminal fragment (MST3/N) translocates to the nucleus and promotes programmed cells death. -

Cerebral Cavernous Malformations: from Genes to Proteins to Disease
See the corresponding editorial in this issue, pp 119–121. J Neurosurg 116:122–132, 2012 Cerebral cavernous malformations: from genes to proteins to disease Clinical article DANIEL D. CAVALCANTI, M.D., M. YASHAR S. KALANI, M.D., PH.D., NIKOLAY L. MARTIROSYAN, M.D., JUSTIN EALES, B.S., ROBERT F. SPETZLER, M.D., AND MARK C. PREUL, M.D. Division of Neurological Surgery, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona Over the past half century molecular biology has led to great advances in our understanding of angio- and vas- culogenesis and in the treatment of malformations resulting from these processes gone awry. Given their sporadic and familial distribution, their developmental and pathological link to capillary telangiectasias, and their observed chromosomal abnormalities, cerebral cavernous malformations (CCMs) are regarded as akin to cancerous growths. Although the exact pathological mechanisms involved in the formation of CCMs are still not well understood, the identification of 3 genetic loci has begun to shed light on key developmental pathways involved in CCM pathogen- esis. Cavernous malformations can occur sporadically or in an autosomal dominant fashion. Familial forms of CCMs have been attributed to mutations at 3 different loci implicated in regulating important processes such as proliferation and differentiation of angiogenic precursors and members of the apoptotic machinery. These processes are important for the generation, maintenance, and pruning of every vessel in the body. In this review the authors highlight the lat- est discoveries pertaining to the molecular genetics of CCMs, highlighting potential new therapeutic targets for the treatment of these lesions.