Genetic and Molecular Dissection of Homocysteinemia in Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Association Analyses of Known Genetic Variants with Gene
ASSOCIATION ANALYSES OF KNOWN GENETIC VARIANTS WITH GENE EXPRESSION IN BRAIN by Viktoriya Strumba A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in The University of Michigan 2009 Doctoral Committee: Professor Margit Burmeister, Chair Professor Huda Akil Professor Brian D. Athey Assistant Professor Zhaohui S. Qin Research Statistician Thomas Blackwell To Sam and Valentina Dmitriy and Elizabeth ii ACKNOWLEDGEMENTS I would like to thank my advisor Professor Margit Burmeister, who tirelessly guided me though seemingly impassable corridors of graduate work. Throughout my thesis writing period she provided sound advice, encouragement and inspiration. Leading by example, her enthusiasm and dedication have been instrumental in my path to becoming a better scientist. I also would like to thank my co-advisor Tom Blackwell. His careful prodding always kept me on my toes and looking for answers, which taught me the depth of careful statistical analysis. His diligence and dedication have been irreplaceable in most difficult of projects. I also would like to thank my other committee members: Huda Akil, Brian Athey and Steve Qin as well as David States. You did not make it easy for me, but I thank you for believing and not giving up. Huda’s eloquence in every subject matter she explained have been particularly inspiring, while both Huda’s and Brian’s valuable advice made the completion of this dissertation possible. I would also like to thank all the members of the Burmeister lab, both past and present: Sandra Villafuerte, Kristine Ito, Cindy Schoen, Karen Majczenko, Ellen Schmidt, Randi Burns, Gang Su, Nan Xiang and Ana Progovac. -

Efficacy and Mechanistic Evaluation of Tic10, a Novel Antitumor Agent
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2012 Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Joshua Edward Allen University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Oncology Commons Recommended Citation Allen, Joshua Edward, "Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent" (2012). Publicly Accessible Penn Dissertations. 488. https://repository.upenn.edu/edissertations/488 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/488 For more information, please contact [email protected]. Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Abstract TNF-related apoptosis-inducing ligand (TRAIL; Apo2L) is an endogenous protein that selectively induces apoptosis in cancer cells and is a critical effector in the immune surveillance of cancer. Recombinant TRAIL and TRAIL-agonist antibodies are in clinical trials for the treatment of solid malignancies due to the cancer-specific cytotoxicity of TRAIL. Recombinant TRAIL has a short serum half-life and both recombinant TRAIL and TRAIL receptor agonist antibodies have a limited capacity to perfuse to tissue compartments such as the brain, limiting their efficacy in certain malignancies. To overcome such limitations, we searched for small molecules capable of inducing the TRAIL gene using a high throughput luciferase reporter gene assay. We selected TRAIL-inducing compound 10 (TIC10) for further study based on its induction of TRAIL at the cell surface and its promising therapeutic index. TIC10 is a potent, stable, and orally active antitumor agent that crosses the blood-brain barrier and transcriptionally induces TRAIL and TRAIL-mediated cell death in a p53-independent manner. -

Meta-Analysis of Nasopharyngeal Carcinoma
BMC Genomics BioMed Central Research article Open Access Meta-analysis of nasopharyngeal carcinoma microarray data explores mechanism of EBV-regulated neoplastic transformation Xia Chen†1,2, Shuang Liang†1, WenLing Zheng1,3, ZhiJun Liao1, Tao Shang1 and WenLi Ma*1 Address: 1Institute of Genetic Engineering, Southern Medical University, Guangzhou, PR China, 2Xiangya Pingkuang associated hospital, Pingxiang, Jiangxi, PR China and 3Southern Genomics Research Center, Guangzhou, Guangdong, PR China Email: Xia Chen - [email protected]; Shuang Liang - [email protected]; WenLing Zheng - [email protected]; ZhiJun Liao - [email protected]; Tao Shang - [email protected]; WenLi Ma* - [email protected] * Corresponding author †Equal contributors Published: 7 July 2008 Received: 16 February 2008 Accepted: 7 July 2008 BMC Genomics 2008, 9:322 doi:10.1186/1471-2164-9-322 This article is available from: http://www.biomedcentral.com/1471-2164/9/322 © 2008 Chen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Epstein-Barr virus (EBV) presumably plays an important role in the pathogenesis of nasopharyngeal carcinoma (NPC), but the molecular mechanism of EBV-dependent neoplastic transformation is not well understood. The combination of bioinformatics with evidences from biological experiments paved a new way to gain more insights into the molecular mechanism of cancer. Results: We profiled gene expression using a meta-analysis approach. Two sets of meta-genes were obtained. Meta-A genes were identified by finding those commonly activated/deactivated upon EBV infection/reactivation. -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

MTHFR) and Methionine Synthase Reductase (MTRR
ANTICANCER RESEARCH 28 : 2807-2812 (2008) Methylenetetrahy drofolate Reductase (MTHFR) and Methionine Synthase Reductase (MTRR) Gene Polymorphisms as Risk Factors for Hepatocellular Carcinoma in a Korean Population SUN YOUNG KWAK 1,2 , UN KYUNG KIM 3, HYO JIN CHO 2, HEE KEUN LEE 3, HYE JIN KIM 2, NAM KEUN KIM 2 and SEONG GYU HWANG 1,2 1Department of Internal Medicine, 2Institute for Clinical Research, College of Medicine, Pochon CHA University, Seongnam; 3Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu, South Korea Abstract. Hepatocellular carcinoma (HCC) is the third were increased risk factors for the disease. The MTHFR most frequent cause of cancer death in South Korea, but 1298A>C and the MTRR 66A>G genotypes were associated genetic susceptibility factors of HCC have not been examined with an increased risk of HCC in th is Korean population. extensively. Methylenetetrahydrofolate reductase (MTHFR) Further studies involving larger and varied populations and methionine synthase reductase (MTRR) play an essential could provide a potential tool for cancer risk assessment in role in both DNA synthesis and methylation and patients who are at risk of developing HCC. polymorphisms in the MTHFR gene, 677C>T, 1298A>C and the MTRR gene, 66A>G, are associated with several types of Hepatocellular carcinoma (HCC) is one of the most common malignancy. In this study, the allelic frequencies and malignant tumors with the third highest mortality rate among genotype distribution of three polymorphisms in the MTHFR malignancies in South Korea. According to a statistical report and MTRR genes from 96 hepatocellular carcinoma (HCC) in 2006, it occurred in 33.8/10,000 men and 11.2/10,000 patients and 201 controls were examined to assess the women in South Korea in 2005 (1). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
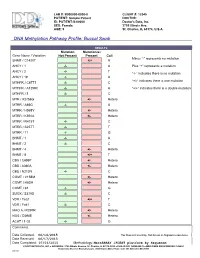
DNA Methylation Pathway Profile; Buccal Swab
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-00000 Doctor's Data, Inc. SEX: Female 3755 Illinois Ave. AGE: 5 St. Charles, IL 60174, U.S.A. !DNA Methylation Pathway Profile; Buccal Swab RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call Minus “-“ represents no mutation SHMT / C1420T +/+ A AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A “+/-“ indicates there is one mutation MTHFR / C677T -/- C MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G -/- A MTRR / H595Y +/- Hetero MTRR / K350A +/- Hetero MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 -/- G BHMT / 1 -/- A BHMT / 2 -/- C BHMT / 4 +/- Hetero BHMT / 8 +/+ T CBS / C699T +/- Hetero CBS / A360A +/- Hetero CBS / N212N -/- C COMT / V158M +/- Hetero COMT / H62H +/- Hetero COMT / 61 -/- G SUOX / S370S -/- C VDR / Taq1 +/+ T VDR / Fok1 -/- C MAO A / R297R +/- Hetero NOS / D298E +/- Hetero ACAT / 1-02 -/- G Comments: Date Collected: 06/14/2015 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 06/17/2015 Date Completed: 07/03/2015 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

DNA Methylation Pathway Profile; Blood Spot
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-000000000 Doctor's Data, Inc. SEX: Male 3755 Illinois Ave AGE: 45 St. Charles, IL 60174 USA !DNA Methylation Pathway Profile; Blood Spot RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call SHMT / C1420T +/- Hetero Minus “-“ represents no mutation AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A MTHFR / C677T -/- C “+/-“ indicates there is one mutation MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G +/- Hetero MTRR / H595Y -/- C MTRR / K350A -/- A MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 +/- Hetero BHMT / 1 -/- A BHMT / 2 +/- Hetero BHMT / 4 +/+ C BHMT / 8 +/+ T CBS / C699T -/- C CBS / A360A -/- C CBS / N212N -/- C COMT / V158M +/+ A COMT / H62H +/+ T COMT / 61 -/- G SUOX / S370S -/- CG VDR / Taq1 -/- C VDR / Fok1 -/- C MAO A / R297R -/- G NOS / D298E -/- G ACAT / 1-02 +/- Hetero Comments: Date Collected: 11/08/2012 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 11/13/2012 Date Completed: 12/10/2012 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

Analysis of Seven Maternal Polymorphisms of Genes Involved In
July 2006 ⅐ Vol. 8 ⅐ No. 7 article Analysis of seven maternal polymorphisms of genes involved in homocysteine/folate metabolism and risk of Down syndrome offspring Iris Scala, MD1, Barbara Granese, PhD1, Maria Sellitto, MD1, Serena Salome`, MD1, Annalidia Sammartino, MD2, Antonio Pepe, PhD1, Pierpaolo Mastroiacovo, MD3, Gianfranco Sebastio, MD1, and Generoso Andria, MD1 PURPOSE: We present a case-control study of seven polymorphisms of six genes involved in homocysteine/folate pathway as risk factors for Down syndrome. Gene-gene/allele-allele interactions, haplotype analysis and the association with age at conception were also evaluated. METHODS: We investigated 94 Down syndrome-mothers and 264 control-women from Campania, Italy. RESULTS: Increased risk of Down syndrome was associated with the methylenetetrahydrofolate reductase (MTHFR) 1298C allele (OR 1.46; 95% CI 1.02–2.10), the MTHFR 1298CC genotype (OR 2.29; 95% CI 1.06–4.96), the reduced-folate-carrier1 (RFC1) 80G allele (1.48; 95% CI 1.05–2.10) and the RFC1 80 GG genotype (OR 2.05; 95% CI 1.03–4.07). Significant associations were found between maternal age at conception Ն34 years and either the MTHFR 1298C or the RFC 180G alleles. Positive interactions were found for the following genotype-pairs: MTHFR 677TT and 1298CC/CA, 1298CC/CA and RFC1 80 GG/GA, RFC1 80 GG and methylenetetrahydrofolate-dehydrogenase 1958 AA. The 677–1298 T-C haplotype at the MTHFR locus was also a risk factor for Down syndrome (P ϭ 0.0022). The methionine-synthase-reductase A66G, the methionine-synthase A2756G and the cystathionine-beta-synthase 844ins68 polymorphisms were not associated with increased risk of Down syndrome. -

Egfr Activates a Taz-Driven Oncogenic Program in Glioblastoma
EGFR ACTIVATES A TAZ-DRIVEN ONCOGENIC PROGRAM IN GLIOBLASTOMA by Minling Gao A thesis submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland March 2020 ©2020 Minling Gao All rights reserved Abstract Hyperactivated EGFR signaling is associated with about 45% of Glioblastoma (GBM), the most aggressive and lethal primary brain tumor in humans. However, the oncogenic transcriptional events driven by EGFR are still incompletely understood. We studied the role of the transcription factor TAZ to better understand master transcriptional regulators in mediating the EGFR signaling pathway in GBM. The transcriptional coactivator with PDZ- binding motif (TAZ) and its paralog gene, the Yes-associated protein (YAP) are two transcriptional co-activators that play important roles in multiple cancer types and are regulated in a context-dependent manner by various upstream signaling pathways, e.g. the Hippo, WNT and GPCR signaling. In GBM cells, TAZ functions as an oncogene that drives mesenchymal transition and radioresistance. This thesis intends to broaden our understanding of EGFR signaling and TAZ regulation in GBM. In patient-derived GBM cell models, EGF induced TAZ and its known gene targets through EGFR and downstream tyrosine kinases (ERK1/2 and STAT3). In GBM cells with EGFRvIII, an EGF-independent and constitutively active mutation, TAZ showed EGF- independent hyperactivation when compared to EGFRvIII-negative cells. These results revealed a novel EGFR-TAZ signaling axis in GBM cells. The second contribution of this thesis is that we performed next-generation sequencing to establish the first genome-wide map of EGF-induced TAZ target genes. -

To See an Example Report
John Doe 02/16/2016 Recommendations based on DNA results – Please follow only under the advice and care of your physician. • Minimize caffeine • Avoid ibuprofen and naproxen regularly and you may need reduced doses of blood thinners if you ever need them • See handout on CYP2D6 for possible drug sensitivities • Avoid GMO foods and high oxalate foods if have been using antibiotics or gut needs healing • Avoid gluten containing foods and supplements. Also consider eliminating dairy and soy • If cholesterol high, watch cholesterol in foods • Check RBC folate and homocysteine levels • Check ammonia and molybdenum levels • Avoid fluoroquinolones, such as Cipro and Levaquin • Eat beets, lamb, and spinach as a source of betaine • Do you have chemical sensitivities to perfume or cigarette smoke? (possible need for SAMe or phosphatidyl choline) • Do you experience anxiety or panic attacks? (betaine, TMG, check RBC folate levels) • If sensitivities to sulfur foods, remove for three days, then introduce back in and see how you feel. Ammonia and molybdenum levels can also be checked for a block in sulfur pathway. If sulfur a problem, stay away from oxalate foods and address possible leaky gut. Supplement Recommendations • Nattoserrazimes (CYP4V2, ITGB3, NR1I12, F11) • Phosphatidyl choline (PEMT; if multiple chemical sensitivities) • Molybdenum (SUOX; check multi vitamin) • Sam-e (AHCY, MTR/MTRR) • TMG (BHMT, but check for anxiety because of COMT) • Quatrefolic (5-MTHF; MTHFR, MTHFD1, MTHFR, MTHFS, MTR/MTRR, PEMT, MAO A) • Probiotic synergy (FUT2) • 5-HTP (MAO A) • PharmaGABA (GAD1) • Adenosyl B12 (COMT, TCN1/2) • Cod liver oil (for vitamin A; BCMO1) • Mitochondrial NRG (NDUFS7) © Ruthieharper MD What You Need to Know In the world of genetic testing, things can become complicated very quickly. -

Stereocilia-Staircase Spacing Is Influenced by Myosin III Motors And
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Special Education and Communication Department of Special Education and Disorders Faculty Publications Communication Disorders 2016 Stereocilia-staircase spacing is influenced yb myosin III motors and their cargos espin-1 and espin-like Seham Ebrahim National Institutes of Health, Bethesda, Maryland Matthew R. Avenarius Oregon Health & Science University, Portland M’hamed Grati National Institutes of Health, Bethesda, Maryland Jocelyn F. Krey Oregon Health & Science University, Portland Alanna M. Windsor National Institutes of Health, Bethesda, Maryland See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/specedfacpub Part of the Special Education and Teaching Commons Ebrahim, Seham; Avenarius, Matthew R.; Grati, M’hamed; Krey, Jocelyn F.; Windsor, Alanna M.; Sousa, Aurea D.; Ballesteros, Angela; Cui, Runjia; Millis, Bryan A.; Salles, Felipe T.; Baird, Michelle A.; Davidson, Michael W.; Jones, Sherri M.; Choi, Dongseok; Dong, Lijin; Raval, Manmeet H.; Yengo, Christopher M.; Gillespie, Peter G. Barr-; and Kachar, Bechara, "Stereocilia-staircase spacing is influenced yb myosin III motors and their cargos espin-1 and espin-like" (2016). Special Education and Communication Disorders Faculty Publications. 100. https://digitalcommons.unl.edu/specedfacpub/100 This Article is brought to you for free and open access by the Department of Special Education and Communication Disorders at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Special Education and Communication Disorders Faculty Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Seham Ebrahim, Matthew R. Avenarius, M’hamed Grati, Jocelyn F. Krey, Alanna M. Windsor, Aurea D.