MSTO1 Mutations Cause Mtdna Depletion, Manifesting As Muscular Dystrophy with Cerebellar Involvement
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mitoxplorer, a Visual Data Mining Platform To
mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, Cecilia Garcia-Perez, José Villaveces, Salma Gamal, Giovanni Cardone, Fabiana Perocchi, et al. To cite this version: Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, et al.. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dy- namics and mutations. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkz1128. hal-02394433 HAL Id: hal-02394433 https://hal-amu.archives-ouvertes.fr/hal-02394433 Submitted on 4 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gkz1128 Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gkz1128/5651332 by Bibliothèque de l'université la Méditerranée user on 04 December 2019 mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim1,†, Prasanna Koti1,†, Adrien Bonnard2, Fabio Marchiano3, Milena Durrbaum¨ 1, Cecilia Garcia-Perez4, Jose Villaveces1, Salma Gamal1, Giovanni Cardone1, Fabiana Perocchi4, Zuzana Storchova1,5 and Bianca H. -

Novel Biallelic Variants in MSTO1 Associated with Mitochondrial Myopathy
Downloaded from molecularcasestudies.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press 1 Novel biallelic variants in MSTO1 associated with mitochondrial myopathy 2 3 Laura Schultz-Rogers1, Alejandro Ferrer1, Nikita R. Dsouza2, Michael T. Zimmermann2,3, Benn 4 E. Smith4, Eric W. Klee1,5, Radhika Dhamija6 5 6 1Center for Individualized Medicine, Mayo Clinic, Rochester, MN, 55905 USA 7 2Bioinformatics Research and Development Laboratory, Genomics Sciences and Precision 8 Medicine Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA 9 3Clinical and Translational Sciences Institute, Medical College of Wisconsin, Milwaukee, WI 10 53226, USA 11 4Department of Neurology, Mayo Clinic, Scottsdale, AZ, 85259 USA 12 5Department of Clinical Genomics, Mayo Clinic, Rochester, MN, 55905 USA 13 6Department of Medical Genetics, Mayo Clinic, Phoenix, AZ, 85054 USA 14 Corresponding author’s email: [email protected] 15 16 HPO TERMS: 17 Hypotonia, early HP:0008947; Restrictive lung disease HP:0002091; Elevated creatine kinase 18 HP:0003236; Delayed gross motor development HP:0002194; Speech articulation difficulties 19 HP:0009088; Dysphagia HP:0002015; Waddling gait HP:0002515; Pectus excavatum 20 HP:0000767; Lumbar kyphosis HP:0008454; EMG: myopathic abnormalities HP:0003458 21 22 SHORT RUNNING TITLE: Novel biallelic variants in MSTO1 myopathy 23 24 25 26 Downloaded from molecularcasestudies.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press 27 ABSTRACT 28 Mitochondrial disorders are caused by nuclear and mitochondrial pathogenic variants 29 leading to defects in mitochondrial function and cellular respiration. Recently, the nuclear- 30 encoded mitochondrial fusion gene MSTO1 (Misato 1) has been implicated in mitochondrial 31 myopathy and ataxia. -

A Study of Alterations in DNA Epigenetic Modifications (5Mc and 5Hmc) and Gene Expression Influenced by Simulated Microgravity I
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Digital Repository @ Iowa State University Genome Informatics Facility Publications Genome Informatics Facility 1-28-2016 A Study of Alterations in DNA Epigenetic Modifications (5mC and 5hmC) and Gene Expression Influenced by Simulated Microgravity in Human Lymphoblastoid Cells Basudev Chowdhury Purdue University Arun S. Seetharam Iowa State University, [email protected] Zhiping Wang Indiana University School of Medicine Yunlong Liu Indiana University School of Medicine Amy C. Lossie Purdue University See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/genomeinformatics_pubs Part of the Bioinformatics Commons, Genetics Commons, and the Genomics Commons Recommended Citation Chowdhury, Basudev; Seetharam, Arun S.; Wang, Zhiping; Liu, Yunlong; Lossie, Amy C.; Thimmapuram, Jyothi; and Irudayaraj, Joseph, "A Study of Alterations in DNA Epigenetic Modifications (5mC and 5hmC) and Gene Expression Influenced by Simulated Microgravity in Human Lymphoblastoid Cells" (2016). Genome Informatics Facility Publications. 4. https://lib.dr.iastate.edu/genomeinformatics_pubs/4 This Article is brought to you for free and open access by the Genome Informatics Facility at Iowa State University Digital Repository. It has been accepted for inclusion in Genome Informatics Facility Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. A Study of Alterations in DNA Epigenetic Modifications (5mC and 5hmC) and Gene Expression Influenced by Simulated Microgravity in Human Lymphoblastoid Cells Abstract Cells alter their gene expression in response to exposure to various environmental changes. Epigenetic mechanisms such as DNA methylation are believed to regulate the alterations in gene expression patterns. -

Novel Biallelic Variants in MSTO1 Associated with Mitochondrial Myopathy
COLD SPRING HARBOR Molecular Case Studies | RESEARCH REPORT Novel biallelic variants in MSTO1 associated with mitochondrial myopathy Laura Schultz-Rogers,1 Alejandro Ferrer,1 Nikita R. Dsouza,2 Michael T. Zimmermann,2,3 Benn E. Smith,4 Eric W. Klee,1,5 and Radhika Dhamija6 1Center for Individualized Medicine, Mayo Clinic, Rochester, Minnesota 55905, USA; 2Bioinformatics Research and Development Laboratory, Genomics Sciences and Precision Medicine Center, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, USA; 3Clinical and Translational Sciences Institute, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, USA; 4Department of Neurology, Mayo Clinic, Scottsdale, Arizona 85259, USA; 5Department of Clinical Genomics, Mayo Clinic, Rochester, Minnesota 55905, USA; 6Department of Medical Genetics, Mayo Clinic, Phoenix, Arizona 85054, USA Abstract Mitochondrial disorders are caused by nuclear and mitochondrial pathogenic variants leading to defects in mitochondrial function and cellular respiration. Recently, the nuclear-encoded mitochondrial fusion gene MSTO1 (Misato 1) has been implicated in mitochondrial myopathy and ataxia. Here we report on a 30-yr-old man presenting with a maternally inherited NM_018116.3:c.651C>G, p.F217L missense variant as well as a paternally inherited arr[GRCh37] 1q22(155581773_155706887) × 1 deletion encompass- ing exons 7–14 of MSTO1. His phenotype included muscle weakness, hypotonia, early mo- tor developmental delay, pectus excavatum, and scoliosis. Testing revealed elevated Corresponding author: plasma creatine kinase, and electromyogram results were consistent with longstanding [email protected] generalized myopathy. These phenotypic features overlap well with previously reported pa- tients harboring biallelic MSTO1 variants. Additionally, our patient presents with dysphagia © 2019 Schultz-Rogers et al. This and restrictive lung disease, not previously reported for MSTO1-associated disorders. -

Transcriptomic Changes Resulting from STK32B Overexpression Identifies Pathways Potentially Relevant to Essential Tremor
bioRxiv preprint doi: https://doi.org/10.1101/552901; this version posted May 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Transcriptomic changes resulting from 2 STK$%B overexpression identifies pathways 3 potentially relevant to essential tremor 4 Calwing Liao.,0, Faezeh Sarayloo.,0, Veikko Vuokila0, Daniel Rochefort0, Fulya Akçimen.,0, 5 Simone Diamond0, Alexandre D. Laporte0, Dan Spiegelman0, Qin HeJ, Hélène Catoire0, Patrick A. 6 Dion0,O, Guy A. Rouleau.,0,O 7 8 .Department of Human GeneticS, McGill University, Montréal, Quebec, Canada 9 0Montreal Neurological Institute, McGill University, Montréal, Quebec, Canada. 10 JDepartment of Biomedical ScienceS, Université de Montréal, Montréal, Quebec, Canada 11 ODepartment of Neurology and Neurosurgery, McGill University, Montréal, Quebec, Canada 12 13 Corresponding Author: 14 Dr. Guy A. Rouleau, MD, PhD, FRCPC, OQ 15 Department of Neurology and Neurosurgery 16 McGill University 17 Montréal, Québec, Canada 18 HJA 0BO 19 E-mail: [email protected] 20 21 Keywords: STK$%B, eSSential tremor, transcriptome, FUS 22 Conflict of Interests: All authors report no conflict of intereStS. 23 Funding Sources: Canadian InstituteS of Health ReSearch 24 bioRxiv preprint doi: https://doi.org/10.1101/552901; this version posted May 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Identification of New Risk Factors for Rolandic Epilepsy: CNV at Xp22.31
Neurogenetics ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmedgenet-2018-105319 on 22 May 2018. Downloaded from Identification of new risk factors for rolandic epilepsy: CNV at Xp22.31 and alterations at cholinergic synapses Laura Addis,1,2 William Sproviero,1 Sanjeev V Thomas,3 Roberto H Caraballo,4 Stephen J Newhouse,5,6,7 Kumudini Gomez,8 Elaine Hughes,9 Maria Kinali,10 David McCormick,9 Siobhan Hannan,10 Silvia Cossu,11,12 Jacqueline Taylor,13 Cigdem I Akman,14 Steven M Wolf,15 David E Mandelbaum,16 Rajesh Gupta,17 Rick A van der Spek,18 Dario Pruna,12 Deb K Pal1 ► Additional material is ABSTRact speech arrest. The characteristic EEG abnormality published online only. To view Background Rolandic epilepsy (RE) is the most is blunt centrotemporal spikes (CTS) typically with please visit the journal online (http:// dx. doi. org/ 10. 1136/ common genetic childhood epilepsy, consisting of focal, frontal positivity and sleep activation. RE starts jmedgenet- 2018- 105319). nocturnal seizures and frequent neurodevelopmental at a mean of 7 years, and there is a spontaneous impairments in speech, language, literacy and attention. remission of seizures during adolescence.3 4 Neuro- For numbered affiliations see A complex genetic aetiology is presumed in most, with developmental comorbidities such as speech sound end of article. monogenic mutations in GRIN2A accounting for >5% of disorder, language impairment, reading disability, cases. migraine and attention impairment are common Correspondence to 5–7 Dr Laura Addis, Department of Objective To identify rare, causal CNV in patients with both in children with RE and their families ; Basic and Clinical Neuroscience, RE. -

The Protective Effect of Club Cell Secretory Protein (CC-16) on COPD Risk And
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.885384; this version posted December 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The protective effect of club cell secretory protein (CC-16) on COPD risk and progression: a Mendelian randomisation study AUTHORS: Stephen Milne1,2,3, Xuan Li1, Ana I Hernandez Cordero1, Chen Xi Yang1, Michael H Cho4, Terri H Beaty5, Ingo Ruczinski6, Nadia N Hansel7, Yohan Bossé8, Corry-Anke Brandsma9, Don D Sin1,2, Ma’en Obeidat1 1. Centre for Heart Lung Innovation, St Paul’s Hospital and University of British Columbia, Vancouver, BC, Canada 2. Division of Respiratory Medicine, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada 3. Faculty of Medicine and Health, University of Sydney, Sydney, New South Wales, Australia 4. Channing Division of Network Medicine and Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Boston, MA, USA 5. Department of Epidemiology, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA 6. Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA 7. Pulmonary and Critical Care Medicine, School of Medicine, Johns Hopkins University, Baltimore, MD, USA 8. Institut universitaire de cardiologie et de pneumologie de Québec, Department of Molecular Medicine, Laval University, Quebec City, Canada 9. University of Groningen Department of Pathology and Medical Biology, University Medical Centre Groningen, Groningen, The Netherlands bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.885384; this version posted December 21, 2019. -

Transcriptional Features of Multiple Myeloma Patients with Chromosome 1Q Gain
Letters to the Editor 1113 References 5 Dworzak MN, Froschl G, Printz D, Mann G, Po¨tschger U, Mu¨hlegger N et al. Prognostic significance and modalities of flow cytometric minimal residual disease detection in childhood acute 1 Woessmann W, Seidemann K, Mann G, Zimmermann M, Burkhardt lymphoblastic leukemia. Blood 2002; 99: 1952–1958. B, Oschlies I et al. The impact of the methotrexate administration 6 Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth schedule and dose in the treatment of children and adolescents with TF et al. A biologic definition of Burkitt’s lymphoma from B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. transcriptional and genomic profiling. N Engl J Med 2006; 354: Blood 2005; 105: 948–958. 2419–2430. 2 Busch K, Borkhardt A, Wossmann W, Reiter A, Harbott J. Combined 7 Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ et al. polymerase chain reaction methods to detect c-myc/IgH rearrange- Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med 2006; ment in childhood Burkitt’s lymphoma for minimal residual disease 354: 2431–2442. analysis. Haematologica 2004; 89: 818–825. 8 Attarbaschi A, Mann G, Dworzak M, Trebo M, Urban C, Fink FM 3 Mussolin L, Basso K, Pillon M, d’Amore ES, Lombardi A, Luzzatto L et al. Malignant non-Hodgkin’s lymphoma of childhood and et al. Prospective analysis of minimal bone marrow infiltration in adolescence in Austria – therapy results between 1986 and 2000. pediatric Burkitt’s lymphomas by long-distance polymerase chain Wien Klin Wochenschr 2002; 114: 978–986. -
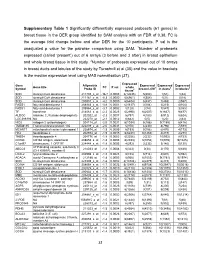
31 Supplementary Table 1 Significantly Differentially Expressed
Supplementary Table 1 Significantly differentially expressed probesets (61 genes) in breast tissue in the DER group identified by SAM analysis with an FDR of 0.38. FC is the average fold change before and after DER for the 10 participants. P val is the unadjusted p value for the pairwise comparison using SAM. 1Number of probesets expressed (called ‘present’) out of 6 arrays (3 before and 3 after) in breast epithelium and whole breast tissue in this study. 2Number of probesets expressed out of 10 arrays in breast ducts and lobules of the study by Turashvili et al (38) and the value in brackets is the median expression level using MAS normalisation (37). Expressed Gene Affymetrix Expressed Expressed Expressed Gene title FC P val whole Symbol Probe ID breast LCM1 in ducts2 in lobules2 tissue1 SCD stearoyl-CoA desaturase 211708_s_at -16.7 0.0003 6(12635) 5(404) 1(51) 1(32) SCD stearoyl-CoA desaturase 211162_x_at -8.3 0.0000 6(6961) 4(354) 1(32) 0(29) SCD stearoyl-CoA desaturase 200831_s_at -4.2 0.0000 6(6404) 5(437) 1(488) 2(557) FADS1 fatty acid desaturase 1 208963_x_at -3.8 0.0001 6(1137) 3(104) 3(215) 4(100) FADS1 fatty acid desaturase 1 208964_s_at -3.7 0.0002 5(128) 2(74) 7(547) 3(355) TF transferrin 214063_s_at -2.3 0.0025 6(2990) 6(2507) 9(1637) 9(877) ALDOC aldolase C, fructose-bisphosphate 202022_at -2.3 0.0007 6(797) 4(183) 8(912) 5(624) LOC389393 NA 222278_at -2.1 0.0012 6(842) 0(5) 1(20) 2(44) NID2 nidogen 2 (osteonidogen) 204114_at -2.0 0.0031 6(1034) 5(186) 9(1188) 6(416) TMEM135 Transmembrane protein 135 222209_s_at -2.0 0.0031 -

Hammoudi Alkaissi
Linköping University Medical Dissertation No.1649 Identification of candidate genes involved in Mercury Toxicokinetics and Mercury Induced Autoimmunity Hammoudi Alkaissi Division of Neuro- and inflammation science, Pathology Department of Clinical and Experimental Medicine Faculty of Medicine and Health Science © Hammoudi Alkaissi, 2018 All rights reserved. Artwork, Cover By – KovCodes Paper I was published in Environmental Health Perspective, with permission of reprint. Paper II was published in PloS One, with permission of reprint. Printed by LiU-Tryck, Linköping, Sweden, 2018 ISBN: 978-91-7685-192-0 ISSN: 0345-0082 You can try but never stop me This is what I'm made of I will never ever let go This is what I'm made of No one can control me Cause this is what I'm made of You can hate but never break me This is what I'm made of Nause – “Made of” (2011) , , . , Main Supervisor Per Hultman Department of Clinical and Experimental Medicine, Linköping University Linköping, Sweden Co-Supervisors Said Havarinasab Department of Clinical and Experimental Medicine, Linköping University. Linköping, Sweden Jesper Bo Nielsen Institute of Public Health, Research Unit for General Practice, University of Southern Denmark. Odense C, Denmark. Peter Söderkvist Department of Clinical and Experimental Medicine, Linköping University. Linköping, Sweden Faculty Opponent Johan Rönnelid Department of Immunology, Genetics and Pathology, Uppsala University. Uppsala, Sweden ABSTRACT BACKGROUND: Autoimmune diseases require the involvement and activation of immune cells and occur when the body builds up an immune response against its own tissues. This process takes place due to the inability to distinguish self-antigen from foreign antigen.