The Effects of Hypoxia on Centrosome Function in Prostate Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information Proximity Interactions Among Centrosome
Current Biology, Volume 24 Supplemental Information Proximity Interactions among Centrosome Components Identify Regulators of Centriole Duplication Elif Nur Firat-Karalar, Navin Rauniyar, John R. Yates III, and Tim Stearns Figure S1 A Myc Streptavidin -tubulin Merge Myc Streptavidin -tubulin Merge BirA*-PLK4 BirA*-CEP63 BirA*- CEP192 BirA*- CEP152 - BirA*-CCDC67 BirA* CEP152 CPAP BirA*- B C Streptavidin PCM1 Merge Myc-BirA* -CEP63 PCM1 -tubulin Merge BirA*- CEP63 DMSO - BirA* CEP63 nocodazole BirA*- CCDC67 Figure S2 A GFP – + – + GFP-CEP152 + – + – Myc-CDK5RAP2 + + + + (225 kDa) Myc-CDK5RAP2 (216 kDa) GFP-CEP152 (27 kDa) GFP Input (5%) IP: GFP B GFP-CEP152 truncation proteins Inputs (5%) IP: GFP kDa 1-7481-10441-1290218-1654749-16541045-16541-7481-10441-1290218-1654749-16541045-1654 250- Myc-CDK5RAP2 150- 150- 100- 75- GFP-CEP152 Figure S3 A B CEP63 – – + – – + GFP CCDC14 KIAA0753 Centrosome + – – + – – GFP-CCDC14 CEP152 binding binding binding targeting – + – – + – GFP-KIAA0753 GFP-KIAA0753 (140 kDa) 1-496 N M C 150- 100- GFP-CCDC14 (115 kDa) 1-424 N M – 136-496 M C – 50- CEP63 (63 kDa) 1-135 N – 37- GFP (27 kDa) 136-424 M – kDa 425-496 C – – Inputs (2%) IP: GFP C GFP-CEP63 truncation proteins D GFP-CEP63 truncation proteins Inputs (5%) IP: GFP Inputs (5%) IP: GFP kDa kDa 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl 1-135136-424425-4961-424136-496FL Ctl Myc- 150- Myc- 100- CCDC14 KIAA0753 100- 100- 75- 75- GFP- GFP- 50- CEP63 50- CEP63 37- 37- Figure S4 A siCtl -

Supplementary Data
SUPPLEMENTARY DATA A cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma Santiago Demajo et al. 1 SUPPLEMENTARY DATA INDEX Supplementary Methods p. 3 Supplementary References p. 8 Supplementary Tables (S1 to S5) p. 9 Supplementary Figures (S1 to S15) p. 17 2 SUPPLEMENTARY METHODS Western blot, immunoprecipitation, and qRT-PCR Western blot (WB) analysis was performed as previously described (1), using cyclin D1 (Santa Cruz Biotechnology, sc-753, RRID:AB_2070433) and tubulin (Sigma-Aldrich, T5168, RRID:AB_477579) antibodies. Co-immunoprecipitation assays were performed as described before (2), using cyclin D1 antibody (Santa Cruz Biotechnology, sc-8396, RRID:AB_627344) or control IgG (Santa Cruz Biotechnology, sc-2025, RRID:AB_737182) followed by protein G- magnetic beads (Invitrogen) incubation and elution with Glycine 100mM pH=2.5. Co-IP experiments were performed within five weeks after cell thawing. Cyclin D1 (Santa Cruz Biotechnology, sc-753), E2F4 (Bethyl, A302-134A, RRID:AB_1720353), FOXM1 (Santa Cruz Biotechnology, sc-502, RRID:AB_631523), and CBP (Santa Cruz Biotechnology, sc-7300, RRID:AB_626817) antibodies were used for WB detection. In figure 1A and supplementary figure S2A, the same blot was probed with cyclin D1 and tubulin antibodies by cutting the membrane. In figure 2H, cyclin D1 and CBP blots correspond to the same membrane while E2F4 and FOXM1 blots correspond to an independent membrane. Image acquisition was performed with ImageQuant LAS 4000 mini (GE Healthcare). Image processing and quantification were performed with Multi Gauge software (Fujifilm). For qRT-PCR analysis, cDNA was generated from 1 µg RNA with qScript cDNA Synthesis kit (Quantabio). qRT–PCR reaction was performed using SYBR green (Roche). -

Molecular Genetics of Microcephaly Primary Hereditary: an Overview
brain sciences Review Molecular Genetics of Microcephaly Primary Hereditary: An Overview Nikistratos Siskos † , Electra Stylianopoulou †, Georgios Skavdis and Maria E. Grigoriou * Department of Molecular Biology & Genetics, Democritus University of Thrace, 68100 Alexandroupolis, Greece; [email protected] (N.S.); [email protected] (E.S.); [email protected] (G.S.) * Correspondence: [email protected] † Equal contribution. Abstract: MicroCephaly Primary Hereditary (MCPH) is a rare congenital neurodevelopmental disorder characterized by a significant reduction of the occipitofrontal head circumference and mild to moderate mental disability. Patients have small brains, though with overall normal architecture; therefore, studying MCPH can reveal not only the pathological mechanisms leading to this condition, but also the mechanisms operating during normal development. MCPH is genetically heterogeneous, with 27 genes listed so far in the Online Mendelian Inheritance in Man (OMIM) database. In this review, we discuss the role of MCPH proteins and delineate the molecular mechanisms and common pathways in which they participate. Keywords: microcephaly; MCPH; MCPH1–MCPH27; molecular genetics; cell cycle 1. Introduction Citation: Siskos, N.; Stylianopoulou, Microcephaly, from the Greek word µικρoκεϕαλi´α (mikrokephalia), meaning small E.; Skavdis, G.; Grigoriou, M.E. head, is a term used to describe a cranium with reduction of the occipitofrontal head circum- Molecular Genetics of Microcephaly ference equal, or more that teo standard deviations -

Centrosome Impairment Causes DNA Replication Stress Through MLK3
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.09.898684; this version posted January 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Centrosome impairment causes DNA replication stress through MLK3/MK2 signaling and R-loop formation Zainab Tayeh 1, Kim Stegmann 1, Antonia Kleeberg 1, Mascha Friedrich 1, Josephine Ann Mun Yee Choo 1, Bernd Wollnik 2, and Matthias Dobbelstein 1* 1) Institute of Molecular Oncology, Göttingen Center of Molecular Biosciences (GZMB), University Medical Center Göttingen, Göttingen, Germany 2) Institute of Human Genetics, University Medical Center Göttingen, Göttingen, Germany *Lead Contact. Correspondence and requests for materials should be addressed to M. D. (e-mail: [email protected]; ORCID 0000-0001-5052-3967) Running title: Centrosome integrity supports DNA replication Key words: Centrosome, CEP152, CCP110, SASS6, CEP152, Polo-like kinase 4 (PLK4), DNA replication, DNA fiber assays, R-loops, MLK3, MK2 alias MAPKAPK2, Seckel syndrome, microcephaly. Highlights: • Centrosome defects cause replication stress independent of mitosis. • MLK3, p38 and MK2 (alias MAPKAPK2) are signalling between centrosome defects and DNA replication stress through R-loop formation. • Patient-derived cells with defective centrosomes display replication stress, whereas inhibition of MK2 restores their DNA replication fork progression and proliferation. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.01.09.898684; this version posted January 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Centriole Overduplication Is the Predominant Mechanism Leading to Centrosome Amplification in Melanoma
Published OnlineFirst January 12, 2018; DOI: 10.1158/1541-7786.MCR-17-0197 Oncogenes and Tumor Suppressors Molecular Cancer Research Centriole Overduplication is the Predominant Mechanism Leading to Centrosome Amplification in Melanoma Ryan A. Denu1,2, Maria Shabbir3, Minakshi Nihal3, Chandra K. Singh3, B. Jack Longley3,4,5, Mark E. Burkard2,4, and Nihal Ahmad3,4,5 Abstract Centrosome amplification (CA) is common in cancer and can evaluated. PLK4 is significantly overexpressed in melanoma com- arise by centriole overduplication or by cell doubling events, pared with benign nevi and in a panel of human melanoma cell including the failure of cell division and cell–cell fusion. To assess lines (A375, Hs294T, G361, WM35, WM115, 451Lu, and SK-MEL- the relative contributions of these two mechanisms, the number of 28) compared with normal human melanocytes. Interestingly, centrosomes with mature/mother centrioles was examined by although PLK4 expression did not correlate with CA in most cases, immunofluorescence in a tissue microarray of human melanomas treatment of melanoma cells with a selective small-molecule PLK4 and benign nevi (n ¼ 79 and 17, respectively). The centrosomal inhibitor (centrinone B) significantly decreased cell proliferation. protein 170 (CEP170) was used to identify centrosomes with The antiproliferative effects of centrinone B were also accompa- mature centrioles; this is expected to be present in most centro- nied by induction of apoptosis. somes with cell doubling, but on fewer centrosomes with over- duplication. Using this method, it was determined that the major- Implications: This study demonstrates that centriole overdupli- ity of CA in melanoma can be attributed to centriole overduplica- cation is the predominant mechanism leading to centrosome tion rather than cell doubling events. -
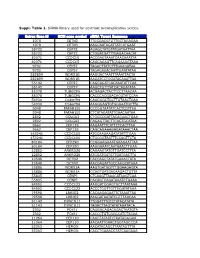
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

A Genome-Scale Protein Interaction Profile of Drosophila P53
A genome-scale protein interaction profile of Drosophila p53 uncovers additional nodes of the human p53 network Andrea Lunardia,b,1, Giulio Di Minina,b,1, Paolo Proveroc, Marco Dal Ferroa,b, Marcello Carottia,b, Giannino Del Sala,b, and Licio Collavina,b,2 aLaboratorio Nazionale Consorzio Interuniversitario per le Biotecnologie (LNCIB), Area Science Park, 34012 Trieste, Italy; bDipartimento di Scienze della Vita, Università degli Studi di Trieste, 34129 Trieste, Italy; and cMolecular Biotechnology Center and Dipartimento di Genetica, Biologia e Biochimica, Università degli Studi di Torino, 10126 Torino, Italy Communicated by Marc W. Kirschner, Harvard Medical School, Boston, MA, March 2, 2010 (received for review June 9, 2009) The genome of the fruitfly Drosophila melanogaster contains a single to better understand the functions of all members of the p53 family p53-like protein, phylogenetically related to the ancestor of the mam- in mammals. malian p53 family of tumor suppressors. We reasoned that a compre- hensive map of the protein interaction profile of Drosophila p53 Results (Dmp53) might help identify conserved interactions of the entire Small Pool in Vitro Expression Cloning (IVEC) Screen for Drosophila p53 family in man. Using a genome-scale in vitro expression cloning p53 Interactors. The Drosophila Gene Collection (DGC) comprises approach, we identified 91 previously unreported Dmp53 interactors, full-length annotated cDNAs of the majority of known genes in considerably expanding the current Drosophila p53 interactome. D. melanogaster (16). Clones from DGC1.0 and DGC2.0 were pu- Looking for evolutionary conservation of these interactions, we rified and pooled in groups of 24. Recombinant maltose binding tested 41 mammalian orthologs and found that 37 bound to one or protein (MBP)-Dmp53 fusion protein was prepared from Bacu- more p53-family members when overexpressed in human cells. -

Regulation of the Centriole Life Cycle by Mdm1 a Dissertation Submitted to the Department of Biology and the Committee on Gradua
REGULATION OF THE CENTRIOLE LIFE CYCLE BY MDM1 A DISSERTATION SUBMITTED TO THE DEPARTMENT OF BIOLOGY AND THE COMMITTEE ON GRADUATE STUDIES OF STANFORD UNIVERSITY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Daniel Patrick Van de Mark August 2014 © 2014 by Daniel Patrick Van de Mark. All Rights Reserved. Re-distributed by Stanford University under license with the author. This work is licensed under a Creative Commons Attribution- Noncommercial 3.0 United States License. http://creativecommons.org/licenses/by-nc/3.0/us/ This dissertation is online at: http://purl.stanford.edu/gf463wx1210 ii I certify that I have read this dissertation and that, in my opinion, it is fully adequate in scope and quality as a dissertation for the degree of Doctor of Philosophy. Tim Stearns, Primary Adviser I certify that I have read this dissertation and that, in my opinion, it is fully adequate in scope and quality as a dissertation for the degree of Doctor of Philosophy. Martha Cyert I certify that I have read this dissertation and that, in my opinion, it is fully adequate in scope and quality as a dissertation for the degree of Doctor of Philosophy. William Nelson I certify that I have read this dissertation and that, in my opinion, it is fully adequate in scope and quality as a dissertation for the degree of Doctor of Philosophy. Rajat Rohatgi Approved for the Stanford University Committee on Graduate Studies. Patricia J. Gumport, Vice Provost for Graduate Education This signature page was generated electronically upon submission of this dissertation in electronic format. -

Strand Breaks for P53 Exon 6 and 8 Among Different Time Course of Folate Depletion Or Repletion in the Rectosigmoid Mucosa
SUPPLEMENTAL FIGURE COLON p53 EXONIC STRAND BREAKS DURING FOLATE DEPLETION-REPLETION INTERVENTION Supplemental Figure Legend Strand breaks for p53 exon 6 and 8 among different time course of folate depletion or repletion in the rectosigmoid mucosa. The input of DNA was controlled by GAPDH. The data is shown as ΔCt after normalized to GAPDH. The higher ΔCt the more strand breaks. The P value is shown in the figure. SUPPLEMENT S1 Genes that were significantly UPREGULATED after folate intervention (by unadjusted paired t-test), list is sorted by P value Gene Symbol Nucleotide P VALUE Description OLFM4 NM_006418 0.0000 Homo sapiens differentially expressed in hematopoietic lineages (GW112) mRNA. FMR1NB NM_152578 0.0000 Homo sapiens hypothetical protein FLJ25736 (FLJ25736) mRNA. IFI6 NM_002038 0.0001 Homo sapiens interferon alpha-inducible protein (clone IFI-6-16) (G1P3) transcript variant 1 mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 15 GALNTL5 NM_145292 0.0001 (GALNT15) mRNA. STIM2 NM_020860 0.0001 Homo sapiens stromal interaction molecule 2 (STIM2) mRNA. ZNF645 NM_152577 0.0002 Homo sapiens hypothetical protein FLJ25735 (FLJ25735) mRNA. ATP12A NM_001676 0.0002 Homo sapiens ATPase H+/K+ transporting nongastric alpha polypeptide (ATP12A) mRNA. U1SNRNPBP NM_007020 0.0003 Homo sapiens U1-snRNP binding protein homolog (U1SNRNPBP) transcript variant 1 mRNA. RNF125 NM_017831 0.0004 Homo sapiens ring finger protein 125 (RNF125) mRNA. FMNL1 NM_005892 0.0004 Homo sapiens formin-like (FMNL) mRNA. ISG15 NM_005101 0.0005 Homo sapiens interferon alpha-inducible protein (clone IFI-15K) (G1P2) mRNA. SLC6A14 NM_007231 0.0005 Homo sapiens solute carrier family 6 (neurotransmitter transporter) member 14 (SLC6A14) mRNA. -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -

Centriole Triplet Microtubules Are Required for Stable Centriole Formation and Inheritance in Human Cells
bioRxiv preprint doi: https://doi.org/10.1101/147975; this version posted June 8, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Centriole triplet microtubules are required for stable centriole 2 formation and inheritance in human cells 3 4 Jennifer T. Wanga, Dong Kongb, Christian R. Hoernerc, Jadranka Loncarekb, Tim 5 Stearnsa,d,e 6 a. Department of Biology, Stanford University, Stanford CA 7 b. Laboratory of Protein Dynamics and Signaling, Center for Cancer Research – 8 Frederick, National Cancer Institute, National Institutes of Health, Frederick MD 9 c. Department of Medicine – Division of Oncology, Stanford School of Medicine, 10 Stanford CA 11 d. Department of Genetics, Stanford School of Medicine, Stanford CA 12 e. Corresponding author 13 14 Summary 15 16 Centrioles are composed of long-lived microtubules arranged in nine triplets. In 17 unicellular eukaryotes, loss of the noncanonical tubulins, delta-tubulin and epsilon- 18 tubulin, result in loss of the triplet microtubule structure. However, the contribution of 19 triplet microtubules to mammalian centriole formation and stability is unknown. Here, we 20 report the first characterization of delta-tubulin and epsilon-tubulin null human cells. 21 Centrioles in cells lacking either delta-tubulin or epsilon-tubulin lack triplet microtubules 22 and fail to undergo centriole maturation. These aberrant centrioles are formed de novo 23 each cell cycle, but are unstable and do not persist to the next cell cycle, leading to a 24 futile cycle of centriole formation and disintegration. -

Supplementary Tables
SUPPLEMENTARY TABLES Supplementary Table 1. Significantly enriched representative GO annotations (biological process) of CDH23 in DLBCL (LinkedOmics). Enrichment Description FDR LeadingEdgeGene ratio ACHE;GPX1;PM20D2;CHIT1;IDUA;PIPOX;RENBP;SDSL;MP O;ALDH2;PRDX1;CTSH;MAOB;OXCT1;MAOA;SULT1A1;C drug 5.91 OMT;CYP1A2;SULT1A2;AMDHD2;GNPDA2;CYP2S1;PAOX; catabolic 1.95 E-04 AMPD3;ALDH3B1;PCK2;NAGK;IL4I1;MT3;GLDC;PCBD1;G process CSH;SMOX;DPYS;AMT;NNT;EPX;OVGP1;IDE;GPX3;NPL;C YP2F1;CYP2B6;COLQ;LPO;CYP4B1. ACTR2;MSH2;NUDT16;PDS5B;DHX9;UCHL5;WDHD1;STU B1;CDK2;UBC;MSH3;SUPT16H;DTL;SIRT1;RAD54B;PMS1; ESCO2;FANCM;RAD21;SFPQ;SLC30A9;USP28;BRIP1;CDC5 L;MCM8;CIB1;FOXM1;DEK;MAGEF1;PRMT6;ATXN3;NON 1.76 DNA repair 1.52 O;LIG3;RMI1;ETAA1;USP43;POLD3;CHEK1;FANCC;PDS5A; E-04 NUCKS1;POLA1;RAD52;CDC7;MPG;RNF169;UBE2N;EXO1; PRKCG;KLHL15;BRCA1;CLSPN;FBXO6;EP300;PAXIP1;SMC 1A;BARD1;NFRKB;DNA2;RNF138;MORF4L2;NIPBL;ASCC1 ;PIAS4;CUL4A;ERCC4;TREX1;USP1; etc. IL18;MMP9;GSN;PLD1;VAMP3;NPC2;ATG7;PSAP;CLU;PYC ARD;FTH1;CDA;VAT1;C8G;ACTR2;CD63;CHIT1;AGPAT2;M AN2B1;TOM1;CTSD;APAF1;PTGES2;BRI3;ITGAX;MSH2;GR N;LAMP1;DPP7;LTA4H;MMP25;VNN1;SERPINA3;C5AR1;CT SA;HEXB;CST3;NCR3;ATAD5;PYGB;GAA;PI4K2A;LTA;ZP3; leukocyte 1.10 ORM2;C1R;C1S;RAC1;SERPINB6;CFP;S100A13;ORM1;ATP6 mediated 1.40 E-04 V0C;TCIRG1;CTSZ;ARSA;TNFRSF1B;ACLY;MVP;PKP1;MA immunity NBA;ASAH1;CD68;RAB24;TMEM173;MPO;C1RL;GM2A;TI MP2;FUCA2;TMEM63A;CHI3L1;PRDX1;CTSH;FTL;EMP2;IT GB2;LRRC7;HLA- F;PLEKHO2;GSTP1;PVR;PRKCZ;CHRNB4;CYFIP1;ITGAM;V AMP2;ANPEP;ANXA2;GALNS;RAB3A;RAB6A; etc.