Infectious Causes of Microcephaly: Epidemiology, Pathogenesis, Diagnosis, And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of the Tetracore Orthopox Biothreat® Antigen Detection Assay
Journal of Virological Methods 187 (2013) 37–42 Contents lists available at SciVerse ScienceDirect Journal of Virological Methods jou rnal homepage: www.elsevier.com/locate/jviromet ® Evaluation of the Tetracore Orthopox BioThreat antigen detection assay using laboratory grown orthopoxviruses and rash illness clinical specimens a,∗ b a a a Michael B. Townsend , Adam MacNeil , Mary G. Reynolds , Christine M. Hughes , Victoria A. Olson , a a Inger K. Damon , Kevin L. Karem a Centers for Disease Control and Prevention, Division of High-Consequence Pathogens and Pathology, Poxvirus and Rabies Branch, 1600 Clifton Road NE, Mailstop G-06, Atlanta, GA 30333, United States b Centers for Disease Control and Prevention, Global Immunization Division, CGH, 1600 Clifton Road NE, Mailstop G-06, Atlanta, GA 30333, United States a b s t r a c t ® Article history: The commercially available Orthopox BioThreat Alert assay for orthopoxvirus (OPV) detection is piloted. Received 5 December 2011 This antibody-based lateral-flow assay labels and captures OPV viral agents to detect their presence. Serial Received in revised form 23 August 2012 dilutions of cultured Vaccinia virus (VACV) and Monkeypox virus (MPXV) were used to evaluate the sensi- Accepted 30 August 2012 tivity of the Tetracore assay by visual and quantitative determinations; specificity was assessed using a Available online 5 September 2012 small but diverse set of diagnostically relevant blinded samples from viral lesions submitted for routine ® OPV diagnostic testing. The BioThreat Alert assay reproducibly detected samples at concentrations of Keywords: 7 6 10 pfu/ml for VACV and MPXV and positively identified samples containing 10 pfu/ml in 4 of 7 inde- Monkeypox Orthopoxvirus pendent experiments. -

Seroprevalence of TORCH Infections in Children
Chattagram Maa-O-Shishu Hospital Medical College Journal Volume 17, Issue 1, January 2018 Original Article Seroprevalence of TORCH Infections in Children Sanjoy Kanti Biswas1* Abstract 2 Md. Badruddoza Background : The acronym “TORCH” was introduced to highlight a group of Nahid Sultana1 pathogens that cause a congenital and perinatal infections: Toxoplasma gondi, rubella virus, Cytomegalovirus (CMV) and Herpes Simplex Virus (HSV). These pathogens are often associated with congenital anomalies. Congenital malformations 1 Department of Microbiology have a direct impact on the family. This study was undertaken to detect the Chattagram Maa-O-Shishu Hospital Medical College Chittagong, Bangladesh. serological evidence of TORCH infections in children, by establishing the presence of specific IgM antibodies. Methods: During the period 1st June 2016 to 30th May 2 Department of Paediatrics 2017, 58 suspected TORCH infection cases were included from Paediatrics Chattagram Maa-O-Shishu Hospital Medical College Chittagong, Bangladesh. Department of CMOSH for TORCH antibody detection. The children were in the age of 0 day to 1 year with an average age of 3.3±2.59 months. The serum samples were tested forIgM and IgG antibodies against TORCH agents by using enzyme linked immunoassay method (ELISA). Results: Among the 58 children, seropositivity was found in 55 (94.82%) cases. Of the 55 seropositive cases serological evidence for combination of IgM and IgG with any one of the TORCH agents was detected in 25 (43.10%) and IgG alone was detected in 30 (51.72%) children. IgM/IgG antibody positivity to Toxoplasma, Rubella, CMV and HSV was 21(36.21%), 50(86.21%), 52(89.66%) and 8(13.79%) respectively. -

Investigation of Poxvirus Host-Range and Gene Expression in Mammalian Cells
ABSTRACT Title: INVESTIGATION OF POXVIRUS HOST-RANGE AND GENE EXPRESSION IN MAMMALIAN CELLS. Jorge David Méndez Ríos, Doctor of Philosophy, 2014. Directed By: Dr. Bernard Moss, Chief, Laboratory of Viral Diseases, NIAID, NIH; Adjunct Professor Department of Cell Biology and Molecular Genetics, University of Maryland; Members of the Poxviridae family have been known as human pathogens for centuries. Their impact in society included several epidemics that decimated the population. In the last few centuries, Smallpox was of great concern that led to the development of our modern vaccines. The systematic study of Poxvirus host-range and immunogenicity provided the knowledge to translate those observations into practice. After the global vaccination campaign by the World Health Organization, Smallpox was the first infectious disease to be eradicated. Nevertheless, diseases such as Monkeypox, Molluscum contagiosum, new bioterrorist threads, and the use of poxviruses as vaccines or vectors provided the necessity to further understand the host-range from a molecular level. Here, we take advantage of the newly developed technologies such as 454 pyrosequencing and RNA-Seq to address previously unresolved questions for the field. First, we were able to identify the Erytrhomelagia-related poxvirus (ERPV) 25 years after its isolation in Hubei, China. Whole-genome sequencing and bioinformatics identified ERPV as an Ectromelia strain closely related to the Ectromelia Naval strain. Second, by using RNA-Seq, the first MOCV in vivo and in vitro transcriptome was generated. New tools have been developed to support future research and for this human pathogen. Finally, deep-sequencing and comparative genomes of several recombinant MVAs (rMVAs) in conjunction with classical virology allowed us to confirm several genes (O1, F5, C17, F11) association to plaque formation in mammalian cell lines. -

Rubella ! PROTOCOL CHECKLIST
Rubella ! PROTOCOL CHECKLIST Enter available information into Merlin upon receipt of initial report, ideally by the next business day Review background on disease, case definition, and laboratory testing Contact health care provider Contact reporting laboratory and request that specimens be sent to the Bureau of Public Health Laboratories (BPHL) for testing Interview patient or guardian Review disease facts Modes of transmission Incubation period Symptoms Ask about exposure to relevant risk factors Immunization history Travel Contact with a known infected or symptomatic person(s) Recent visit to a healthcare setting Identify settings where exposures may have occurred and all known contacts (Sections 6 and 7) Determine evidence of immunity to rubella for contacts Recommend health care provider consultation for exposed women who are pregnant or trying to become pregnant. Monitor contacts for the duration of the incubation period Determine whether patient, symptomatic contacts, or susceptible contacts have exposures in sensitive situation (e.g., school, child care, college dormitory, military, other congregate living settings, health care workers, etc.) Ensure isolation of symptomatic contacts Identify those at-risk with unknown immune status (susceptible persons) for vaccination as indicated Provide education on prevention through vaccination Address patient’s questions or concerns Follow-up on special situations, including persons in sensitive situations and pregnant women Enter additional data obtained from interview into Merlin Rubella Guide to Surveillance and Investigation Rubella 1. DISEASE REPORTING A. Purpose of reporting and surveillance 1. To prevent congenital rubella syndrome (CRS). 2. To assure that children with suspected CRS are tested to confirm or rule out the diagnosis in a timely manner, to assure prompt treatment, and prevent spread of the disease. -

German Measles
Rubella (German Measles) Summary Rubella is an infectious viral disease characterized by mild clinical disease, where cases are often subclinical, when symptomatic individuals may present with an erythematous maculopapular rash, lymphadenopathy and a low-grade fever. Infection with the rubella virus causes two distinct illnesses: congenital rubella syndrome (CRS) and postnatal rubella. Rubella virus occurs worldwide. It is most prevalent in winter and spring. In the United States, rubella has been largely controlled after the advent of immunization. The incidence of rubella in the U.S. has decreased by approximately 99% from the pre-vaccine era. Epidemic rubella in the U.S. last occurred in 1964. Agent Rubella virus is in the Togaviridae family, genus Rubivirus. Transmission Reservoir: Humans. Mode of transmission: For postnatal rubella, direct or droplet contact with nasopharyngeal secretions of infected persons. Infants with CRS may shed virus in nasopharyngeal secretions and urine for one year or more and can transmit infection to susceptible contacts. Period of communicability: A few days to 7 days after the onset of rash. Infants with CRS may shed virus in nasopharyngeal secretions and urine for one year or more and can transmit infection to susceptible contacts. Clinical Disease Incubation period: For postnatally acquired rubella, usually 16-18 days; range 14-21 days. Illness: Postnatal rubella is usually a mild disease with diffuse erythematous maculopapular rash, lymphademopathy (commonly sub-occipital, postauricular and cervical) and fever. Adults sometimes have a prodromal illness of headache, malaise, coryza, and conjunctivitis. Arthralgias and arthritis can frequently complicate postnatal rubella, especially in females. Leukopenia and thrombocytopenia can occur, but hemorrhagic complications are rare. -

Measles, Mumps, Rubella, Varicella Jul 2020
Measles, Mumps, Rubella, Varicella Jul 2020 Health Care Professional Programs Measles, mumps, rubella and varicella are vaccine-preventable diseases. The efficacy of two doses of vaccine (one for rubella) is close to 100% for measles, 76-95% for mumps, 95% for rubella, and 98-100% for varicella. If born before 1970, you may be immune to measles, mumps and rubella due to naturally acquired infection; after 1970 you most likely received one or two vaccines. You may be immune to varicella due to naturally acquired infection or you may have received one or two vaccines (vaccine introduced in Canada in 1999). If you are unable to locate your vaccination records, revaccination is safe unless you are pregnant or immunocompromised. Measles: Measles is one of the most highly communicable infectious diseases with greater than 90% secondary attack rates among susceptible persons. Symptoms include fever, cough, runny nose, red eyes, Koplik spots (white spots on the inner lining of the mouth), followed by a rash that begins on the face, advances to the trunk and then to the arms and legs. The virus is transmitted by the airborne route, respiratory droplets, or direct contact with nasal or throat secretions of infected persons. The incubation period is 7 to 18 days. Cases are infectious from 4 days before the beginning of the prodromal period to 4 days after rash onset. Mumps: Mumps virus is highly contagious and is transmitted primarily by droplet spread, as well as by direct contact with saliva of an infected person. Symptoms of mumps virus infection include fever, headache and muscle aches followed by swelling in one or more salivary glands (usually parotid gland). -

Involvement of a P53-Dependent Pathway in Rubella Virus-Induced Apoptosis
Virology 259, 74–84 (1999) Article ID viro.1999.9757, available online at http://www.idealibrary.com on Involvement of a p53-Dependent Pathway in Rubella Virus-Induced Apoptosis Klara Megyeri,*,† Klara Berencsi,* Thanos D. Halazonetis,* George C. Prendergast,* Giorgia Gri,* Stanley A. Plotkin,‡ Giovanni Rovera,* and Eva Go¨nczo¨l*,†,1 *The Wistar Institute, 3601 Spruce Street, Philadelphia, Pennsylvania 19104; †Department of Microbiology, Albert Szent-Gyo¨rgyi Medical University, Dom ter 10, Szeged, H-6720, Hungary; and ‡Pasteur Merieux Connaught, 4650 Wismer Road, Doylestown, Pennsylvania 18901 Received October 6, 1998; returned to author for revision December 8, 1998; accepted April 1, 1999 In light of the important role of apoptotic cell death in the pathogenesis of several viral infections, we asked whether the cytopathogenicity evoked by rubella virus (RV) might also involve apoptotic mechanisms. The To-336 strain of RV induced apoptosis in Vero and RK-13 cells, but not in fibroblast cell lines. UV-inactivated RV virions did not elicit the apoptotic response, indicating that productive infection is required for the induction of cell death. Both p53 and p21 protein levels were highly elevated in RV-infected Vero cells. The level of p21 mRNA was increased, while expression of the p53 gene was unaffected by RV infection. A dominant-negative p53 mutant (p53W248) conferred partial protection from RV-induced apoptosis. These data implicate a p53-dependent apoptotic pathway in the cytopathogenicity of RV, thereby suggesting a mechanism by which RV exerts its teratogenic effects. © 1999 Academic Press INTRODUCTION activation of the Fas and tumor necrosis factor-receptor signaling pathways (Wada et al., 1995; Takizawa et al., Viruses rely on cellular metabolites and enzymes for 1996; Diaz-Guerra et al., 1997; Lee et al., 1997; Su and their replication and perturb many strictly monitored bio- Schneider, 1997). -

Ev20n1p53.Pdf (663.1Kb)
SEROLOGIC SCREENING FOR CYTOMEGALOVIRUS, RUBELLA VIRUS, HERPESSIMPLEX VIRUS, HEPATITIS B VIRUS, AND Z-DXOl?~snIlA GONDII IN TWO URBAN POPULATIONS OF PREGNANT WOMEN IN CHILE1 Pablo Vzlzl;2 Jorge Toves-Pereyra, 3 Sergio Stagno, 4 Francisco Gonzhfez,’ Enrique Donoso, G Chades A. Allford, 7 Zimara Hirsch, 8 and Luk Rodtiguezg I NTRODUCTION these agents represent naturally acquired infections. There are only a few reports Although the prevalence of concerning the epidemiology of congeni- congenital and perinatal infections is tal and perinatal infections in Chile (l- high, however, it is.unclear whether their $&In general, these reports show that incidence during the childbearing years the prevalences of infection with most of (and hence their potential to cause fetal the causative agents are high and that disease) is different from that observed in most infections are acquired at an early communities such as those in developed age. Since vaccinations against cytomeg- countries where lower prevalences are the alovirus (CMV), herpes simplex virus rule. Therefore, in order to help assess (HSV), and Toxopl’asma gondii have not the importance of CMV, HSV, rubella, yet been introduced, high prevalences of HBV, and ToxopLasmagondii as causesof z 2 < ’ This article is also being published in Spanish in the 4 Department of Pediatrics, University of Alabama. Bir- 2 BoLefin de /a Oficina Sanitankz Panameticana, 99(5), mingham. Alabama, United States. % 1985. The work reported here was supported by Grant s Division of Obstetrics and Gynecology, Dr. Sotero de1 x ‘4 LHZ503Q from the Pan American Health Organita- Rio Hospital, Puente Alto, Chile. u 6 Department of Obstetrics and Gynecology. -
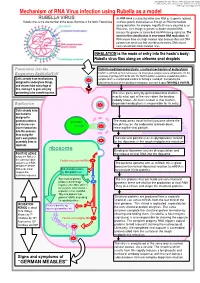
Mechanism of RNA Virus Infection Using Rubella As a Model
put together by Alex Yartsev: Sorry if i used your images or data and forgot to reference you. Tell me who you are. [email protected] Mechanism of RNA Virus infection using Rubella as a model An RNA virus is a virus that either uses RNA as its genetic material, Rubella virus is the sole member of the genus Rubivirus in the family Togaviridae or whose genetic material passes through an RNA intermediate during replication. For example, Hepatitis B virus is classified as an RNA virus, even though its genome is double-stranded DNA, because the genome is transcribed into RNA during replication. The basis for this classification is error-prone RNA replication: All RNA viruses have very high mutation rates because they lack DNA polymerases which can find and edit out mistakes. DNA viruses have considerably lower mutation rates. INHALATION is the mode of entry into the hosts’s body: Rubella virus flies along on airborne snot droplets Penetration into the Clathrin-mediated endocytosis: a normal mechanism of endocytosis Respiratory EpithelialCell Clathrin is common as mud; nerves use it to endocytose synaptic vesicle components, it’s the usual way of getting stuff into the cell. The Clathrin protein assembles a basket-like protein Cells already have mechanisms complex which is a complicated machine for forming a “coated pit”, manipulating the designed to endocytose things, cytoskeleton to involute the lipid bilayer membrane, and close it again forming a vesicle. and viruses take advantage of this, fooling it to gain entry by pretending to be something else. The virus gains entry by getting bound to clathrin. -

Rubella Virus
Kingdom of Saudi Arabia King Saud University College of Science ]اكتب نصاً[ 2016 - 1437 Rubella virus Kingdom of Saudi Arabia King Saud University College of Science Rubella Virus (German measles) Prepared by: Bashayer Aseeri Reem Abahussain Safiah Al-mushawah Norah Al-samih Nadia Alruji Supervised by: Norah al-kubaisi 1 Rubella virus Table of Contents Introduction ........................................................................................................................ 3 A.History of the Disease .................................................................................................. 3 B.Introduction to the Virus……. ....................................................................................... 4 C. The distribution of this disease. .................................................................................. 4 D.Epidemic. ...................................................................................................................... 5 Classification of the virus ................................................................................................... 5 Structure and Genome ...................................................................................................... 6 A.Shape ............................................................................................................................ 6 B.Size ............................................................................................................................... 6 C.Envelope ...................................................................................................................... -

Herpes Simplex, Varicella-Zoster, Cytomegalo Virus, and Measles Virus in Patients with Connective Tissue Diseases
An,ii. rheitm. Dis. (I1972), 31, 192 Ann Rheum Dis: first published as 10.1136/ard.31.3.192 on 1 May 1972. Downloaded from Antibody levels to parainfluenza, herpes simplex, varicella-zoster, cytomegalo virus, and measles virus in patients with connective tissue diseases J. L. KALLIOMAKI AND PEKKA HALONEN From the Departmetit of Medicinie anid the Departmenit of Virology, Uniiversity of Tuirkii, Tzrh-ki,, Finlanid Phillips and Christian (1969) reported that measles Subjects and methods antibody titres are higher in patients with systemic in The serum specimens were collected froni 48 consecutive lupus erythematosus (SLE) than controls. Later patients with connective tissue diseases. The number (1970) they also found increased levels of antibody of patients in each diagnostic subgroup is shown in titres to parainfluenza type I virus in patients with Table I. In 26 patients RA was classical or definite; SLE and in six patients with Reiter's syndrome, but one also had dermatitis herpetiformis and foul- with copyright. the mean titres of patients with rheumatoid arthritis classical disease had chronic nephropathy. The SLE (RA) were not significantly different than those of patients had been treated several times at our hospital normal subjects. and the diagnosis was confirmed. The postinfective The present report describes the antibody titres to arthritis group contained four patients with Yelrsiniia parainfluenza type 1, herpes simplex, varicella-zoster, enterocolitica infection and one with salmonellosis, and virus and measles virus in 48 in one patient the disease developed after an influenza- cytomegalo patients like disease. with connective tissue diseases and in 48 normal The serum specimens of the control subjects were the http://ard.bmj.com/ subjects. -

Maternal Immunoreactivity to Herpes Simplex Virus 2 and Risk of Autism Spectrum Disorder in Male Offspring
RESEARCH ARTICLE Clinical Science and Epidemiology crossm Maternal Immunoreactivity to Herpes Simplex Virus 2 and Risk of Autism Spectrum Disorder in Male Offspring Milada Mahic,a,b Siri Mjaaland,a,b Hege Marie Bøvelstad,b Nina Gunnes,b Downloaded from Ezra Susser,c,d Michaeline Bresnahan,c,d Anne-Siri Øyen,b,h Bruce Levin,e Xiaoyu Che,a Deborah Hirtz,f* Ted Reichborn-Kjennerud,b,g Synnve Schjølberg,b Christine Roth,b,h Per Magnus,b Camilla Stoltenberg,b,i Pål Surén,b Mady Hornig,a,c W. Ian Lipkina,c Center for Infection and Immunity, Mailman School of Public Health, Columbia University, New York, New York, USAa; Norwegian Institute of Public Health, Oslo, Norwayb; Department of Epidemiology, Mailman School of Public Health, Columbia University, New York, New York, USAc; New York State Psychiatric Institute, Columbia University, New York, New York, USAd; Department of Biostatistics, Mailman School of Public Health, http://msphere.asm.org/ Columbia University, New York, New York, USAe; National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USAf; Institute of Clinical Medicine, University of Oslo, Oslo, Norwayg; Nic Waals Institute, Lovisenberg Hospital, Oslo, Norwayh; Department of Global Public Health and Primary Care, University of Bergen, Bergen, Norwayi ABSTRACT Maternal infections during pregnancy are associated with risk of neuro- Received 11 January 2017 Accepted 2 developmental disorders, including autism spectrum disorders (ASDs). Proposed February 2017 Published 22 February 2017 pathogenetic mechanisms include fetal infection, placental inflammation, and mater- Citation Mahic M, Mjaaland S, Bøvelstad HM, Gunnes N, Susser E, Bresnahan M, Øyen A-S, nal cytokines or antibodies that cross the placenta.