New Antiepileptic Drugs
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Kava (Piper Methysticum) and Its Methysticin Constituents Protect Brain Tissue Against Ischemic Damage in Rodents
5 Refs: Arletti R et al, Stimulating property of Turnera diffusa and Pfaffia paniculata extracts on the sexual-behavior of male rats. Psychopharmacology 143(1), 15-19, 1999. Berger F, Handbuch der drogenkunde . Vol 2, Maudrich, Wien, 1950. Martinez M, Les plantas medicinales de Mexico . Cuarta Edicion Botas Mexico , p119, 1959. Tyler VE et al, Pharmacognosy , 9 th edition, Lea & Febiger, Philadelphia, 1988. KAVA ( Piper methysticum ) - A REVIEW The Kava plant (Piper methysticum) is a robust, well-branching and erect perennial shrub belonging to the pepper family (Piperaceae). The botanical origin remains unknown, although it is likely that early Polynesian explorers brought the plant with them from island to island. Numerous varieties of Kava exist, and today it is widely cultivated in several Pacific Island countries both for local use as well as the rapidly growing demand for pharmaceutical preparations. The dried rhizomes (roots) are normally used. The first description to the western world of the ceremonial use of an intoxicating beverage prepared from Kava was made by Captain James Cook following his Pacific voyage in 1768. The drink, prepared as an infusion in an elaborate manner after first chewing the root, is consumed on formal occasions or meetings of village elders and chiefs, as well as in reconciling with enemies and on a more social basis. It remains an important social custom in many Pacific Island countries today. Most of the islands of the Pacific possessed Kava prior to European contact, particularly those encompassed by Polynesia, Melanesia and Micronesia. After drinking the Kava beverage a pleasantly relaxed and sociable state develops, after which a deep and restful sleep occurs. -

Alternative Treatments for Depression and Anxiety
2019 PCB Conference: Strickland Benzodiazepines (BZDs), Herbal and Alternative Treatments for Anxiety & Depression BZD Learning Objectives • List at least three uses for benzodiazepines • Discuss at least two risk factors associated with benzodiazepine prescriptions Craig Strickland, PhD, Owner Biobehavioral Education and Consultation https://sites.google.com/site/bioedcon 1 2 BZD Pharmacokinetics Clinical Uses of BZDs Generic Name Trade Name Rapidity ½ Life Dose (mg) • Treat a variety of anxiety disorders alprazolam Xanax Intermediate Short 0.75-4 • Hypnotics • Muscle relaxants chlordiaze- Librium Intermediate Long 15-100 poxide • To produce anterograde amnesia clonazepam Klonopin Intermediate Long 0.5-4 • Alcohol & other CNS depressant withdrawal • Anti-convulsant therapy diazepam Valium Rapid Long 4-40 triazolam Halcion Intermediate Very short 0.125-0.5 temazepam Restoril Short Short 7.5-30 3 4 1 2019 PCB Conference: Strickland Issues with BZDs Herbal Medication and Alternative Therapies Used in the Treatment of Depression and Anxiety • Addictive potential • Confusion between “anti-anxiety” effects and the “warm-fuzzy) • Large dose ranges • Comparison of BZDs with medications like Buspar, etc. • They work, they work well and they work quickly 5 6 Alternative Tx. Learning Objectives Background Information on herbals: Natural does not necessarily mean “safe” • List several amino acid treatments for depression • Side-effects and adverse reactions • List at least three of the most common herbal – Herbal medications are “drugs” although -

(12) Patent Application Publication (10) Pub. No.: US 2004/0224012 A1 Suvanprakorn Et Al
US 2004O224012A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0224012 A1 Suvanprakorn et al. (43) Pub. Date: Nov. 11, 2004 (54) TOPICAL APPLICATION AND METHODS Related U.S. Application Data FOR ADMINISTRATION OF ACTIVE AGENTS USING LIPOSOME MACRO-BEADS (63) Continuation-in-part of application No. 10/264,205, filed on Oct. 3, 2002. (76) Inventors: Pichit Suvanprakorn, Bangkok (TH); (60) Provisional application No. 60/327,643, filed on Oct. Tanusin Ploysangam, Bangkok (TH); 5, 2001. Lerson Tanasugarn, Bangkok (TH); Suwalee Chandrkrachang, Bangkok Publication Classification (TH); Nardo Zaias, Miami Beach, FL (US) (51) Int. CI.7. A61K 9/127; A61K 9/14 (52) U.S. Cl. ............................................ 424/450; 424/489 Correspondence Address: (57) ABSTRACT Eric G. Masamori 6520 Ridgewood Drive A topical application and methods for administration of Castro Valley, CA 94.552 (US) active agents encapsulated within non-permeable macro beads to enable a wider range of delivery vehicles, to provide longer product shelf-life, to allow multiple active (21) Appl. No.: 10/864,149 agents within the composition, to allow the controlled use of the active agents, to provide protected and designable release features and to provide visual inspection for damage (22) Filed: Jun. 9, 2004 and inconsistency. US 2004/0224012 A1 Nov. 11, 2004 TOPCAL APPLICATION AND METHODS FOR 0006 Various limitations on the shelf-life and use of ADMINISTRATION OF ACTIVE AGENTS USING liposome compounds exist due to the relatively fragile LPOSOME MACRO-BEADS nature of liposomes. Major problems encountered during liposome drug Storage in vesicular Suspension are the chemi CROSS REFERENCE TO OTHER cal alterations of the lipoSome compounds, Such as phos APPLICATIONS pholipids, cholesterols, ceramides, leading to potentially toxic degradation of the products, leakage of the drug from 0001) This application claims the benefit of U.S. -

Benzodiazepines: Uses and Risks Charlie Reznikoff, MD Hennepin Healthcare
Benzodiazepines: Uses and Risks Charlie Reznikoff, MD Hennepin healthcare 4/22/2020 Overview benzodiazepines • Examples of benzos and benzo like drugs • Indications for benzos • Pharmacology of benzos • Side effects and contraindications • Benzo withdrawal • Benzo tapers 12/06/2018 Sedative/Hypnotics • Benzodiazepines • Alcohol • Z-drugs (Benzo-like sleeping aids) • Barbiturates • GHB • Propofol • Some inhalants • Gabapentin? Pregabalin? 12/06/2018 Examples of benzodiazepines • Midazolam (Versed) • Triazolam (Halcion) • Alprazolam (Xanax) • Lorazepam (Ativan) • Temazepam (Restoril) • Oxazepam (Serax) • Clonazepam (Klonopin) • Diazepam (Valium) • Chlordiazepoxide (Librium) 4/22/2020 Sedatives: gaba stimulating drugs have incomplete “cross tolerance” 12/06/2018 Effects from sedative (Benzo) use • Euphoria/bliss • Suppresses seizures • Amnesia • Muscle relaxation • Clumsiness, visio-spatial impairment • Sleep inducing • Respiratory suppression • Anxiolysis/disinhibition 12/06/2018 Tolerance to benzo effects? • Effects quickly diminish with repeated use (weeks) • Euphoria/bliss • Suppresses seizures • Effects incompletely diminish with repeated use • Amnesia • Muscle relaxation • Clumsiness, visio-spatial impairment • Seep inducing • Durable effects with repeated use • Respiratory suppression • Anxiolysis/disinhibition 12/06/2018 If you understand this pharmacology you can figure out the rest... • Potency • 1 mg diazepam <<< 1 mg alprazolam • Duration of action • Half life differences • Onset of action • Euphoria, clinical utility in acute -
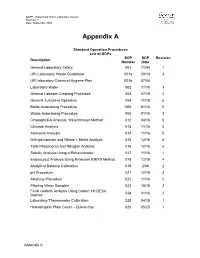
Lab Standard Operating Procedures (Sops)
QAPP –Watershed Watch Laboratory Assays Revision: 7 Date: September 2020 Appendix A Standard Operation Procedures List of SOPs SOP SOP Revision Description Number Date General Laboratory Safety 001 11/04 1 URI Laboratory Waste Guidebook 001a 09/13 3 URI laboratory Chemical Hygiene Plan 001b 07/04 Laboratory Water 002 11/16 3 General Labware Cleaning Procedure 003 07/19 4 General Autoclave Operation 004 10/18 6 Bottle Autoclaving Procedure 005 01/16 5 Waste Autoclaving Procedure 006 01/16 3 Chlorophyll-A Analysis, Welschmeyer Method 012 04/18 6 Chloride Analysis 013 11/16 5 Ammonia Analysis 014 11/16 5 Orthophosphate and Nitrate + Nitrite Analysis 015 12/16 5 Total Phosphorus and Nitrogen Analysis 016 12/16 5 Salinity Analysis Using a Refractometer 017 11/16 1 Enterococci Analysis Using Enterolert IDEXX Method 018 12/19 4 Analytical Balance Calibration 019 2/06 2 pH Procedure 021 12/19 3 Alkalinity Procedure 022 11/16 2 Filtering Water Samples 023 10/18 2 Fecal coliform Analysis Using Colilert 18 IDEXX 024 01/16 2 Method Laboratory Thermometer Calibration 025 04/19 1 Heterotrophic Plate Count – Quanti-tray 026 05/20 1 Appendix A Standard Operating Procedure 001 Date: 11/04 General Laboratory Safety Revision: 1 Author: Linda Green University of Rhode Island Watershed Watch 1.0 PURPOSE AND DESCRIPTION LAB SAFETY IS EVERYBODY’S JOB! Please be sure to familiarize yourself with these general procedures, as well as the specific handling requirements included in the Standard Operating Procedure (SOP) for each analysis/process. Further general information regarding University of Rhode Island standards for health and safety are found in SOP 001a – University Safety and Waste Handling Document. -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

1-(4-Amino-Cyclohexyl)
(19) & (11) EP 1 598 339 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07D 211/04 (2006.01) C07D 211/06 (2006.01) 24.06.2009 Bulletin 2009/26 C07D 235/24 (2006.01) C07D 413/04 (2006.01) C07D 235/26 (2006.01) C07D 401/04 (2006.01) (2006.01) (2006.01) (21) Application number: 05014116.7 C07D 401/06 C07D 403/04 C07D 403/06 (2006.01) A61K 31/44 (2006.01) A61K 31/48 (2006.01) A61K 31/415 (2006.01) (22) Date of filing: 18.04.2002 A61K 31/445 (2006.01) A61P 25/04 (2006.01) (54) 1-(4-AMINO-CYCLOHEXYL)-1,3-DIHYDRO-2H-BENZIMIDAZOLE-2-ONE DERIVATIVES AND RELATED COMPOUNDS AS NOCICEPTIN ANALOGS AND ORL1 LIGANDS FOR THE TREATMENT OF PAIN 1-(4-AMINO-CYCLOHEXYL)-1,3-DIHYDRO-2H-BENZIMIDAZOLE-2-ON DERIVATE UND VERWANDTE VERBINDUNGEN ALS NOCICEPTIN ANALOGE UND ORL1 LIGANDEN ZUR BEHANDLUNG VON SCHMERZ DERIVÉS DE LA 1-(4-AMINO-CYCLOHEXYL)-1,3-DIHYDRO-2H-BENZIMIDAZOLE-2-ONE ET COMPOSÉS SIMILAIRES POUR L’UTILISATION COMME ANALOGUES DU NOCICEPTIN ET LIGANDES DU ORL1 POUR LE TRAITEMENT DE LA DOULEUR (84) Designated Contracting States: • Victory, Sam AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU Oak Ridge, NC 27310 (US) MC NL PT SE TR • Whitehead, John Designated Extension States: Newtown, PA 18940 (US) AL LT LV MK RO SI (74) Representative: Maiwald, Walter (30) Priority: 18.04.2001 US 284666 P Maiwald Patentanwalts GmbH 18.04.2001 US 284667 P Elisenhof 18.04.2001 US 284668 P Elisenstrasse 3 18.04.2001 US 284669 P 80335 München (DE) (43) Date of publication of application: (56) References cited: 23.11.2005 Bulletin 2005/47 EP-A- 0 636 614 EP-A- 0 990 653 EP-A- 1 142 587 WO-A-00/06545 (62) Document number(s) of the earlier application(s) in WO-A-00/08013 WO-A-01/05770 accordance with Art. -

Supplementary Information
Supplementary Information Network-based Drug Repurposing for Novel Coronavirus 2019-nCoV Yadi Zhou1,#, Yuan Hou1,#, Jiayu Shen1, Yin Huang1, William Martin1, Feixiong Cheng1-3,* 1Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA 2Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH 44195, USA 3Case Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA #Equal contribution *Correspondence to: Feixiong Cheng, PhD Lerner Research Institute Cleveland Clinic Tel: +1-216-444-7654; Fax: +1-216-636-0009 Email: [email protected] Supplementary Table S1. Genome information of 15 coronaviruses used for phylogenetic analyses. Supplementary Table S2. Protein sequence identities across 5 protein regions in 15 coronaviruses. Supplementary Table S3. HCoV-associated host proteins with references. Supplementary Table S4. Repurposable drugs predicted by network-based approaches. Supplementary Table S5. Network proximity results for 2,938 drugs against pan-human coronavirus (CoV) and individual CoVs. Supplementary Table S6. Network-predicted drug combinations for all the drug pairs from the top 16 high-confidence repurposable drugs. 1 Supplementary Table S1. Genome information of 15 coronaviruses used for phylogenetic analyses. GenBank ID Coronavirus Identity % Host Location discovered MN908947 2019-nCoV[Wuhan-Hu-1] 100 Human China MN938384 2019-nCoV[HKU-SZ-002a] 99.99 Human China MN975262 -

Deliberate Self-Poisoning with a Lethal Dose of Pentobarbital: Survival with Supportive Care
Deliberate self-poisoning with a lethal dose of pentobarbital: Survival with supportive care. (1) (1,2) Santosh Gone , Andis Graudins (1) Clinical Toxicology Service, Program of Emergency Medicine, Monash Health (2) Monash Emergency Research Collaboration, Clinical Sciences at Monash Health, Monash University Abstract 84 INTRODUCTION CASE REPORT (continued) DISCUSSION Pentobarbital (Nembutal) is short acting In the ED: Pentobarbital has been a banned substance for barbiturate sedative-hypnotic, currently widely GCS: 3/15, fixed dilated pupils, apnoeic and human use in Australia since 1998. However, it can used in veterinary practice for anesthesia and ventilated. be procured overseas or bought on the internet. euthanasia. Pulse: 116 bpm sinus tachycardia BP: 115/60 on epinephrine infusion. Pentobarbital is recommended as an effective It is also commonly recommended as a VBG: pH 7.03 pCO2 77 mmHg Bicarb 19 mmol/L agent for use in euthanasia due to its apparently euthanasia drug for assisted suicide and it is Lactate 8.8 mmol/L peaceful transition to death. unlikely that any resuscitative measures will be Activated charcoal (50g) given via an NG tube. attempted in such cases. Course in the Intensive Care Department: In a case series of 150 assisted suicides in Day-1 post-OD: Sweden, a 100% success rate was seen with Intentional overdose results in depression of - Absent brain stem reflexes and fixed dilated pupils for ingestion of 9 grams. Cardiovascular collapse was brain stem function and rapid onset respiratory five days. seen within 15 minutes in 30% of patients. It is rare depression and apnoea. Hypotension also - Diabetes insipidus developed with urine output of to see survival after intentional ingestion for develops, followed by cardiovascular collapse 300ml/hour. -

(12) Patent Application Publication (10) Pub. No.: US 2010/014.3507 A1 Gant Et Al
US 2010.0143507A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/014.3507 A1 Gant et al. (43) Pub. Date: Jun. 10, 2010 (54) CARBOXYLIC ACID INHIBITORS OF Publication Classification HISTONE DEACETYLASE, GABA (51) Int. Cl. TRANSAMINASE AND SODIUM CHANNEL A633/00 (2006.01) A 6LX 3/553 (2006.01) A 6LX 3/553 (2006.01) (75) Inventors: Thomas G. Gant, Carlsbad, CA A63L/352 (2006.01) (US); Sepehr Sarshar, Cardiff by A6II 3/19 (2006.01) the Sea, CA (US) C07C 53/128 (2006.01) A6IP 25/06 (2006.01) A6IP 25/08 (2006.01) Correspondence Address: A6IP 25/18 (2006.01) GLOBAL PATENT GROUP - APX (52) U.S. Cl. .................... 424/722:514/211.13: 514/221; 10411 Clayton Road, Suite 304 514/456; 514/557; 562/512 ST. LOUIS, MO 63131 (US) (57) ABSTRACT Assignee: AUSPEX The present invention relates to new carboxylic acid inhibi (73) tors of histone deacetylase, GABA transaminase, and/or PHARMACEUTICALS, INC., Sodium channel activity, pharmaceutical compositions Vista, CA (US) thereof, and methods of use thereof. (21) Appl. No.: 12/632,507 Formula I (22) Filed: Dec. 7, 2009 Related U.S. Application Data (60) Provisional application No. 61/121,024, filed on Dec. 9, 2008. US 2010/014.3507 A1 Jun. 10, 2010 CARBOXYLIC ACID INHIBITORS OF HISTONE DEACETYLASE, GABA TRANSAMNASE AND SODIUM CHANNEL 0001. This application claims the benefit of priority of Valproic acid U.S. provisional application No. 61/121,024, filed Dec. 9, 2008, the disclosure of which is hereby incorporated by ref 0004 Valproic acid is extensively metabolised via erence as if written herein in its entirety. -

Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on Β-Alanine and Isatin
Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on β-Alanine and Isatin by Robert Philip Colaguori A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Pharmaceutical Sciences University of Toronto © Copyright by Robert Philip Colaguori, 2016 ii Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on β-Alanine and Isatin Robert Philip Colaguori Master of Science Department of Pharmaceutical Sciences University of Toronto 2016 Abstract Epilepsy is the fourth-most common neurological disorder in the world. Approximately 70% of cases can be controlled with therapeutics, however 30% remain pharmacoresistant. There is no cure for the disorder, and patients affected are subsequently medicated for life. Thus, there is a need to develop compounds that can treat not only the symptoms, but also delay/prevent progression. Previous work resulted in the discovery of NC-2505, a substituted β-alanine with activity against chemically induced seizures. Several N- and α-substituted derivatives of this compound were synthesized and evaluated in the kindling model and 4-AP model of epilepsy. In the kindling model, RC1-080 and RC1-102 were able to decrease the mean seizure score from 5 to 3 in aged mice. RC1-085 decreased the interevent interval by a factor of 2 in the 4-AP model. Future studies are focused on the synthesis of further compounds to gain insight on structure necessary for activity. iii Acknowledgments First and foremost, I would like to thank my supervisor Dr. Donald Weaver for allowing me to join the lab as a graduate student and perform the work ultimately resulting in this thesis. -

Pharmacology on Your Palms CLASSIFICATION of the DRUGS
Pharmacology on your palms CLASSIFICATION OF THE DRUGS DRUGS FROM DRUGS AFFECTING THE ORGANS CHEMOTHERAPEUTIC DIFFERENT DRUGS AFFECTING THE NERVOUS SYSTEM AND TISSUES DRUGS PHARMACOLOGICAL GROUPS Drugs affecting peripheral Antitumor drugs Drugs affecting the cardiovascular Antimicrobial, antiviral, Drugs affecting the nervous system Antiallergic drugs system antiparasitic drugs central nervous system Drugs affecting the sensory Antidotes nerve endings Cardiac glycosides Antibiotics CNS DEPRESSANTS (AFFECTING THE Antihypertensive drugs Sulfonamides Analgesics (opioid, AFFERENT INNERVATION) Antianginal drugs Antituberculous drugs analgesics-antipyretics, Antiarrhythmic drugs Antihelminthic drugs NSAIDs) Local anaesthetics Antihyperlipidemic drugs Antifungal drugs Sedative and hypnotic Coating drugs Spasmolytics Antiviral drugs drugs Adsorbents Drugs affecting the excretory system Antimalarial drugs Tranquilizers Astringents Diuretics Antisyphilitic drugs Neuroleptics Expectorants Drugs affecting the hemopoietic system Antiseptics Anticonvulsants Irritant drugs Drugs affecting blood coagulation Disinfectants Antiparkinsonian drugs Drugs affecting peripheral Drugs affecting erythro- and leukopoiesis General anaesthetics neurotransmitter processes Drugs affecting the digestive system CNS STIMULANTS (AFFECTING THE Anorectic drugs Psychomotor stimulants EFFERENT PART OF THE Bitter stuffs. Drugs for replacement therapy Analeptics NERVOUS SYSTEM) Antiacid drugs Antidepressants Direct-acting-cholinomimetics Antiulcer drugs Nootropics (Cognitive