10 Molecular Regulation of Cardiogenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Evolving Cardiac Lymphatic Vasculature in Development, Repair and Regeneration
REVIEWS The evolving cardiac lymphatic vasculature in development, repair and regeneration Konstantinos Klaourakis 1,2, Joaquim M. Vieira 1,2,3 ✉ and Paul R. Riley 1,2,3 ✉ Abstract | The lymphatic vasculature has an essential role in maintaining normal fluid balance in tissues and modulating the inflammatory response to injury or pathogens. Disruption of normal development or function of lymphatic vessels can have severe consequences. In the heart, reduced lymphatic function can lead to myocardial oedema and persistent inflammation. Macrophages, which are phagocytic cells of the innate immune system, contribute to cardiac development and to fibrotic repair and regeneration of cardiac tissue after myocardial infarction. In this Review, we discuss the cardiac lymphatic vasculature with a focus on developments over the past 5 years arising from the study of mammalian and zebrafish model organisms. In addition, we examine the interplay between the cardiac lymphatics and macrophages during fibrotic repair and regeneration after myocardial infarction. Finally, we discuss the therapeutic potential of targeting the cardiac lymphatic network to regulate immune cell content and alleviate inflammation in patients with ischaemic heart disease. The circulatory system of vertebrates is composed of two after MI. In this Review, we summarize the current complementary vasculatures, the blood and lymphatic knowledge on the development, structure and function vascular systems1. The blood vasculature is a closed sys- of the cardiac lymphatic vasculature, with an emphasis tem responsible for transporting gases, fluids, nutrients, on breakthroughs over the past 5 years in the study of metabolites and cells to the tissues2. This extravasation of cardiac lymphatic heterogeneity in mice and zebrafish. -
Development of Right Ventricle
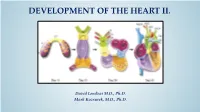
DEVELOPMENT OF THE HEART II. David Lendvai M.D., Ph.D. Mark Kozsurek, M.D., Ph.D. • Septation of the common atrioventricular (AV) orifice. • Formation of the interatrial septum. • Formation of the muscular interventricular septum. • Appearance of the membranous interventricular septum and the spiral aorticopulmonary septum. right left septum primum septum primum septum primum septum primum septum primum septum primum foramen primum foramen primum septum primum septum primum foramen primum foramen primum septum primum septum primum foramen secundum foramen secundum foramen primum foramen primum septum primum foramen secundum septum primum foramen secundum foramen primum foramen primum septum primum septum primum foramen secundum foramen secundum septum secundum septum secundum foramen secundum foramen ovale foramen ovale septum primum septum primum septum secundum septum secundum foramen secundum foramen ovale foramen ovale septum primum septum primum septum secundum septum secundum foramen secundum septum primum foramen ovale foramen ovale septum primum SUMMARY • The septation of the common atrium starts with the appearance of the crescent-shaped septum primum. The opening of this septum, the foramen primum, becomes progressively smaller. • Before the foramen primum completly closes, postero-superiorly several small openings appear on the septum primum. These perforations coalesce later and form the foramen secundum. • On the right side of the septum primum a new septum, the septum secundum, starts to grow. The orifice of the septum secundum is the foramen ovale. • Finally two crescent-like, incomplete, partially overlapping septa exist with one hole on each. Septum secundum is more rigid and the septum primum on its left side acts as a valve letting the blood flow exclusively from the right to the left. -

Abnormalities of Placental Development and Function Are
bioRxiv preprint doi: https://doi.org/10.1101/388074; this version posted August 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abnormalities of placental development and function are associated with the different fetal growth patterns of hypoplastic left heart syndrome and transposition of the great arteries. Weston Troja1, Kathryn J. Owens1, Jennifer Courtney1, Andrea C. Hinton3, Robert B. Hinton3, James F. Cnota2 and Helen N. Jones1* 1. Center for Fetal and Placental Therapy, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, Cincinnati, Ohio 2. Heart Institute, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, Cincinnati, Ohio 3. TriHealth Maternal Fetal Medicine, 375 Dixsmyth Ave, Cincinnati Ohio *Corresponding Author: Helen N. Jones [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/388074; this version posted August 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Background: Birthweight is a critical predictor of congenital heart disease (CHD) surgical outcomes. Hypoplastic left heart syndrome (HLHS) is cyanotic CHD with known fetal growth restriction and placental abnormalities. Transposition of the great arteries (TGA) is cyanotic CHD with normal fetal growth. Comparison of the placenta in these diagnoses may provide insights on the fetal growth abnormality of CHD. Methods: Clinical data and placental histology from placentas associated with Transposition of the Great Arteries (TGA) were analyzed for gross pathology, morphology, maturity and vascularity and compared to both control and previously analyzed HLHS placentas [1]. -

Conditional Mutation of Hand1 in the Mouse Placenta Disrupts Placental Vascular Development Resulting in Fetal Loss in Both Early and Late Pregnancy
International Journal of Molecular Sciences Article Conditional Mutation of Hand1 in the Mouse Placenta Disrupts Placental Vascular Development Resulting in Fetal Loss in Both Early and Late Pregnancy Jennifer A. Courtney 1, Rebecca L. Wilson 2,3, James Cnota 4 and Helen N. Jones 2,3,* 1 Center for Fetal and Placental Therapy, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA; [email protected] 2 Center for Research in Perinatal Outcomes, College of Medicine, University of Florida, Gainesville, FL 32603, USA; rebecca.wilson@ufl.edu 3 Department of Physiology and Functional Genomics, College of Medicine, University of Florida, Gainesville, FL 32603, USA 4 Heart Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA; [email protected] * Correspondence: jonesh@ufl.edu; Tel.: +1-352-846-1503 Abstract: Congenital heart defects (CHD) affect approximately 1% of all live births, and often require complex surgeries at birth. We have previously demonstrated abnormal placental vascularization in human placentas from fetuses diagnosed with CHD. Hand1 has roles in both heart and placen- tal development and is implicated in CHD development. We utilized two conditionally activated Hand1A126fs/+ murine mutant models to investigate the importance of cell-specific Hand1 on placental development in early (Nkx2-5Cre) and late (Cdh5Cre) pregnancy. Embryonic lethality occurred in Citation: Courtney, J.A.; Wilson, R.L.; Nkx2-5Cre/Hand1A126fs/+ embryos with marked fetal demise occurring after E10.5 due to a failure Cnota, J.; Jones, H.N. Conditional in placental labyrinth formation and therefore the inability to switch to hemotrophic nutrition or Mutation of Hand1 in the Mouse Placenta Disrupts Placental Vascular maintain sufficient oxygen transfer to the fetus. -

Functional Morphology of the Cardiac Jelly in the Tubular Heart of Vertebrate Embryos
Review Functional Morphology of the Cardiac Jelly in the Tubular Heart of Vertebrate Embryos Jörg Männer 1,*,† and Talat Mesud Yelbuz 2,† 1 Group Cardio‐Embryology, Institute of Anatomy and Embryology UMG, Georg‐August‐University Goettingen, D‐37075 Goettingen, Germany; [email protected] 2 Department of Cardiac Sciences, King Abdulaziz Cardiac Center, Section of Pediatric Cardiology, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh 11426, Saudi Arabia; [email protected] * Correspondence: [email protected]; Tel.: +49‐551‐39‐7032 † This work is dedicated to the memory of our academic mentors Gerd Steding (1936–2011) and Armin Wessel (1946–2011). Received: 29 January 2019; Accepted: 21 February 2019; Published: 27 February 2019 Abstract: The early embryonic heart is a multi‐layered tube consisting of (1) an outer myocardial tube; (2) an inner endocardial tube; and (3) an extracellular matrix layer interposed between the myocardium and endocardium, called “cardiac jelly” (CJ). During the past decades, research on CJ has mainly focused on its molecular and cellular biological aspects. This review focuses on the morphological and biomechanical aspects of CJ. Special attention is given to (1) the spatial distribution and fiber architecture of CJ; (2) the morphological dynamics of CJ during the cardiac cycle; and (3) the removal/remodeling of CJ during advanced heart looping stages, which leads to the formation of ventricular trabeculations and endocardial cushions. CJ acts as a hydraulic skeleton, displaying striking structural and functional similarities with the mesoglea of jellyfish. CJ not only represents a filler substance, facilitating end‐systolic occlusion of the embryonic heart lumen. -

Conserved Signaling Mechanisms in Drosophila Heart Development
DEVELOPMENTAL DYNAMICS 246:641–656, 2017 DOI: 10.1002/DVDY.24530 REVIEWS Conserved Signaling Mechanisms in Drosophila Heart Development a Shaad M. Ahmad 1,2* 1Department of Biology, Indiana State University, Terre Haute, Indiana 2The Center for Genomic Advocacy, Indiana State University, Terre Haute, Indiana Abstract: Signal transduction through multiple distinct pathways regulates and orchestrates the numerous biological pro- cesses comprising heart development. This review outlines the roles of the FGFR, EGFR, Wnt, BMP, Notch, Hedgehog, Slit/ Robo, and other signaling pathways during four sequential phases of Drosophila cardiogenesis—mesoderm migration, cardiac mesoderm establishment, differentiation of the cardiac mesoderm into distinct cardiac cell types, and morphogenesis of the heart and its lumen based on the proper positioning and cell shape changes of these differentiated cardiac cells—and illus- trates how these same cardiogenic roles are conserved in vertebrates. Mechanisms bringing about the regulation and combi- natorial integration of these diverse signaling pathways in Drosophila are also described. This synopsis of our present state of knowledge of conserved signaling pathways in Drosophila cardiogenesis and the means by which it was acquired should facili- tate our understanding of and investigations into related processes in vertebrates. Developmental Dynamics 246:641–656, 2017. VC 2017 Wiley Periodicals, Inc. Key words: heart development; Drosophila cardiogenesis; cardiac mesoderm specification; cardiac morphogenesis; -

Cardiogenesis in the Bovine to 35 Somites
CARDIOGENESIS IN THE BOVINE TO 35 SOMITES by PATRICIA ANN NODEN A. A., Chanute Junior College, 1962 B. S., Kansas State University, 1964 A MASTER'S THESIS submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE Department of Zoology KANSAS STATE UNIVERSITY Manhattan, Kansas 1966 Approved by: ii N7G> TABLE OF CONTENTS Oocu mtA t INTRODUCTION 1 METHODS AND MATERIALS 6 OBSERVATIONS 9 Presomite Stage 9 Six Somite Stage 9 Nine Somite Stage 10 13 Somite Stage 18 19 Somite Stage 18 22 Somite Stage 19 24-25 Somite Stage 20 28-30 Somite Stage ....... 21 33-35 Somite Stage 24 DISCUSSION 27 CONCLUSION S3 ACKNOWLEDGMENTS 35 BIBLIOGRAPHY . 36 . INTRODUCTION Mammalian cardiogenesis is a vast field which so far has not been thoroughly explored. There are few species in which the complete development of the heart has been studied and many in which partial formation has been observed The formation of the heart to the functional stage in the dog (14 somites) was studied by Bonnet (1901) and Duffey (1953). Duffey's thesis was used exclusively for comparison in this paper. The rat has been studied in some detail, from presomite to birth and after, by Burlingame and Long (1939) and early stages by Ravn (1895) and Goss (1942, 1952). The tubular phase of cardiogenesis in the rabbit of 10-13 somites was described by Girgis (1930, 1933). Dwinnel (1939) worked with early somite stages. Development of the aortic arches was described by Bremer (1912). Hensen (1875), Bom (1888, 1889), Strahl and Carius (1889) and Rouviere (1904) have described early cardiac formation. -

Functional Morphology of the Cardiac Jelly in the Tubular
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 30 January 2019 doi:10.20944/preprints201901.0312.v1 Peer-reviewed version available at J. Cardiovasc. Dev. Dis. 2019, 6, 12; doi:10.3390/jcdd6010012 1 Review 2 Functional Morphology of the Cardiac Jelly in the 3 Tubular Heart of Vertebrate Embryos 4 Jörg Männer 1,* and Talat Mesud Yelbuz 2 5 1 Group Cardio-Embryology, Institute of Anatomy and Embryology UMG, Georg-August-University 6 Goettingen, D-37075 Goettingen, Germany; [email protected] 7 2 Department of Cardiac Sciences, King Abdulaziz Cardiac Center, Section of Pediatric Cardiology, King 8 Abdulaziz Medical City, Ministry of National Guard Health Affairs; Riyadh, Kingdom of Saudi Arabia; 9 [email protected] 10 * Correspondence: [email protected]; Tel.: +49-551-39-7032 11 12 Abstract: The early embryonic heart is a multi-layered tube consisting of (1) an 13 outer myocardial tube; (2) an inner endocardial tube; and (3) an extracellular 14 matrix layer interposed between myocardium and endocardium, called “cardiac 15 jelly” (CJ). During the past decades, research on CJ has mainly focused on its 16 molecular and cell biological aspects. This review focuses on the morphological 17 and biomechanical aspects of CJ. Special attention is given to (1) the spatial 18 distribution and fiber architecture of CJ; (2) the morphological dynamics of CJ 19 during the cardiac cycle; and (3) the removal/remodeling of CJ during advanced 20 heart looping stages, which leads to the formation of ventricular trabeculations 21 and endocardial cushions. CJ acts as a hydraulic skeleton displaying striking 22 structural and functional similarities with the mesoglea of jellyfish. -

6. Heart and Circulatory System I
6. HEART AND CIRCULATORY SYSTEM I Dr. Taube P. Rothman P&S 12-520 [email protected] 212-305-7930 RECOMMENDED READING: Larsen Human Embryology, 3rd Edition, pp. 195-199; 157-169 top left; 172-174; bottom 181-182; 187-top 189, Simbryo-cardiovascular system SUMMARY: The circulatory system, consisting of heart, blood vessels, and blood cells is the first functional organ to develop. This lecture will focus on the formation of the embryonic vasculature, the origin and formation of the early heart tube and primitive cardiac chambers, cardiac looping, and the primitive circulation. Between the 5th - 8th week of embryonic development, the tubular heart is remodeled into a four chambered structure. We will see how right and left atrioventricular canals connect each atrium with its respective ventricle, and how the atrial septum and definitive right and left atria form. We will also see why the great veins deliver blood to the right atrium while the pulmonary veins empty into the left. GLOSSARY: Angioblasts: precursors of blood vessels Angiogenesis: lengthening, branching, sprouting and remodeling of embryonic blood vessels Aortic arches: paired arteries surrounding the pharynx; portions will contribute to formation of the great arterial vessels Blood islands: clusters of cells in the yolk sac, connecting stalk and chorionic villi that form primitive blood vessels Cardiac jelly: gelatinous extracellular matrix that forms the middle layer of the heart tube Ductus venosus: shunts most of the blood in the umbilical vein into the inferior vena cava -

Cardiovascular System - Accessscience from Mcgraw-Hill Education
Cardiovascular system - AccessScience from McGraw-Hill Education http://accessscience.com/content/109900 (http://accessscience.com/) Article by: Weichert, Charles K. College of Arts and Sciences, University of Cincinnati, Cincinnati, Ohio. Copenhaver, W. M. College of Physicians and Surgeons, Columbia University, New York; Department of Biological Structures, School of Medicine, University of Miami, Miami, Florida. Ebert, James D. Department of Embryology, Carnegie Institution, Washington, DC. Patten, Bradley M. Department of Anatomy, University of Michigan, Ann Arbor, Michigan. Jones, David R. Department of Zoology, University of British Columbia, Vancouver, Canada. Publication year: 2014 DOI: http://dx.doi.org/10.1036/1097-8542.109900 (http://dx.doi.org/10.1036/1097-8542.109900) Content Comparative Anatomy Embryogenesis of blood vessels Balancing ventricular output Heart Angiogenesis Human Postnatal Circulation Arterial system Circulatory system morphogenesis Pulmonary circuit and ductus Venous system Primitive venous system Physiological aspects of transition Comparative Embryology Functional Development of Heart Comparative Physiology Heart Contractions of the heart General physiology of circulation Tubular heart formation Heart-forming areas Microcirculation Cardiac loop and regional development Contractile proteins Heart Formation of definitive heart Synthesis of contractile proteins Arteries Partitioning of mammalian heart Action of inhibitors Venous system Division of atrium and ventricles Human Fetal Circulation at Term Bibliography -

Early Patterning and Specification of Cardiac Progenitors in Gastrulating
RESEARCH ARTICLE elifesciences.org Early patterning and specification of cardiac progenitors in gastrulating mesoderm W Patrick Devine1,2,3,5*, Joshua D Wythe1,2, Matthew George1,2,4, Kazuko Koshiba-Takeuchi1,2, Benoit G Bruneau1,2,3,4* 1Gladstone Institute of Cardiovascular Disease, San Francisco, United States; 2Roddenberry Center for Stem Cell Biology and Medicine at Gladstone, San Francisco, United States; 3Cardiovascular Research Institute, University of California, San Francisco, San Francisco, United States; 4Developmental and Stem Cell Biology Program, University of San Francisco, San Francisco, United States; 5Department of Pathology, University of California, San Francisco, San Francisco, United States Abstract Mammalian heart development requires precise allocation of cardiac progenitors. The existence of a multipotent progenitor for all anatomic and cellular components of the heart has been predicted but its identity and contribution to the two cardiac progenitor ‘fields’ has remained undefined. Here we show, using clonal genetic fate mapping, that Mesp1+ cells in gastrulating mesoderm are rapidly specified into committed cardiac precursors fated for distinct anatomic regions of the heart. We identify Smarcd3 as a marker of early specified cardiac precursors and identify within these precursors a compartment boundary at the future junction of the left and right ventricles that arises prior to morphogenesis. Our studies define the timing and hierarchy of cardiac progenitor specification and demonstrate that the cellular and anatomical fate of *For correspondence: patrick. mesoderm-derived cardiac cells is specified very early. These findings will be important to [email protected] understand the basis of congenital heart defects and to derive cardiac regeneration strategies. (WPD); bbruneau@gladstone. -

The Lymphatic System in Zebrafish Heart Development, Regeneration
Journal of Cardiovascular Development and Disease Review The Lymphatic System in Zebrafish Heart Development, Regeneration and Disease Modeling Xidi Feng 1,†, Stanislao Travisano 1,† , Caroline A. Pearson 2, Ching-Ling Lien 1,3,4,* and Michael R. M. Harrison 5,6,* 1 The Saban Research Institute of Children’s Hospital Los Angeles, Los Angeles, CA 90027, USA; [email protected] (X.F.); [email protected] (S.T.) 2 Laboratory of Neurogenetics and Development, Brain and Mind Research Institute, Weill Cornell Medical College, New York, NY 10021, USA; [email protected] 3 Department of Surgery, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA 4 Department of Biochemistry & Molecular Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA 5 Cardiovascular Research Institute, Weill Cornell Medical College, New York, NY 10021, USA 6 Department of Cell and Developmental Biology, Weill Cornell Medical College, New York, NY 10021, USA * Correspondence: [email protected] (C.-L.L.); [email protected] (M.R.M.H.) † These authors contributed equally to this work. Abstract: Heart disease remains the single largest cause of death in developed countries, and novel therapeutic interventions are desperately needed to alleviate this growing burden. The cardiac lymphatic system is the long-overlooked counterpart of the coronary blood vasculature, but its important roles in homeostasis and disease are becoming increasingly apparent. Recently, the car- diac lymphatic vasculature in zebrafish has been described and its role in supporting the potent regenerative response of zebrafish heart tissue investigated. In this review, we discuss these findings in the wider context of lymphatic development, evolution and the promise of this system to open Citation: Feng, X.; Travisano, S.; new therapeutic avenues to treat myocardial infarction and other cardiopathologies.