An Experimental and Theoretical Study of the Valence Shell Photoelectron Spectrum of Bromochlorofluoromethane
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
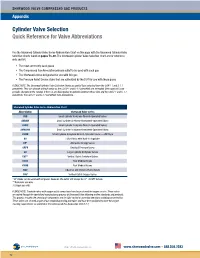
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Evolution of Refrigerants Mark O
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jced Review (R)Evolution of Refrigerants Mark O. McLinden* and Marcia L. Huber Cite This: J. Chem. Eng. Data 2020, 65, 4176−4193 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: As we enter the “fourth generation” of refrigerants, we consider the evolution of refrigerant molecules, the ever- changing constraints and regulations that have driven the need to consider new molecules, and the advancements in the tools and property models used to identify new molecules and design equipment using them. These separate aspects are intimately intertwined and have been in more-or-less continuous development since the earliest days of mechanical refrigeration, even if sometimes out-of-sight of the mainstream refrigeration industry. We highlight three separate, comprehensive searches for new refrigerantsin the 1920s, the 1980s, and the 2010sthat sometimes identified new molecules, but more often, validated alternatives already under consideration. A recurrent theme is that there is little that is truly new. Most of the “new” refrigerants, from R-12 in the 1930s to R- 1234yf in the early 2000s, were reported in the chemical literature decades before they were considered as refrigerants. The search for new refrigerants continued through the 1990s even as the hydrofluorocarbons (HFCs) were becoming the dominant refrigerants in commercial use. This included a return to several long-known natural refrigerants. Finally, we review the evolution of the NIST REFPROP database for the calculation of refrigerant properties. -

"Fluorine Compounds, Organic," In: Ullmann's Encyclopedia Of
Article No : a11_349 Fluorine Compounds, Organic GU¨ NTER SIEGEMUND, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany WERNER SCHWERTFEGER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany ANDREW FEIRING, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States BRUCE SMART, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States FRED BEHR, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States HERWARD VOGEL, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota, United States BLAINE MCKUSICK, E. I. DuPont de Nemours & Co., Wilmington, Delaware, United States 1. Introduction....................... 444 8. Fluorinated Carboxylic Acids and 2. Production Processes ................ 445 Fluorinated Alkanesulfonic Acids ...... 470 2.1. Substitution of Hydrogen............. 445 8.1. Fluorinated Carboxylic Acids ......... 470 2.2. Halogen – Fluorine Exchange ......... 446 8.1.1. Fluorinated Acetic Acids .............. 470 2.3. Synthesis from Fluorinated Synthons ... 447 8.1.2. Long-Chain Perfluorocarboxylic Acids .... 470 2.4. Addition of Hydrogen Fluoride to 8.1.3. Fluorinated Dicarboxylic Acids ......... 472 Unsaturated Bonds ................. 447 8.1.4. Tetrafluoroethylene – Perfluorovinyl Ether 2.5. Miscellaneous Methods .............. 447 Copolymers with Carboxylic Acid Groups . 472 2.6. Purification and Analysis ............. 447 8.2. Fluorinated Alkanesulfonic Acids ...... 472 3. Fluorinated Alkanes................. 448 8.2.1. Perfluoroalkanesulfonic Acids -

THOMAS MIDGLEY, JR., and the INVENTION of CHLOROFLUOROCARBON REFRIGERANTS: IT AIN’T NECESSARILY SO Carmen J
66 Bull. Hist. Chem., VOLUME 31, Number 2 (2006) THOMAS MIDGLEY, JR., AND THE INVENTION OF CHLOROFLUOROCARBON REFRIGERANTS: IT AIN’T NECESSARILY SO Carmen J. Giunta, Le Moyne College The 75th anniversary of the first public Critical readers are well aware description of chlorofluorocarbon (CFC) of the importance of evaluating refrigerants was observed in 2005. A sources of information. For ex- symposium at a national meeting of ample, an article in Nature at the the American Chemical Society (ACS) end of 2005 tested the accuracy on CFCs from invention to phase-out of two encyclopedias’ entries on a (1) and an article on their invention sample of topics about science and and inventor in the Chemical Educa- history of science (3). A thought- tor (2) marked the occasion. The story ful commentary published soon of CFCs—their obscure early days as afterwards raised questions on just laboratory curiosities, their commercial what should count as an error in debut as refrigerants, their expansion into assessing such articles: omissions? other applications, and the much later disagreements among generally re- discovery of their deleterious effects on liable sources (4)? Readers of his- stratospheric ozone—is a fascinating torical narratives who are neither one of science and society well worth practicing historians nor scholarly telling. That is not the purpose of this amateurs (the category to which I article, though. aspire) may find an account of a Thomas Midgley, Jr. historical research process attrac- This paper is about contradictory Courtesy Richard P. Scharchburg tive. As a starting point, consider sources, foggy memories, the propaga- Archives, Kettering University the following thumbnail summary tion of error, and other obstacles to writ- of the invention. -

Overview of Fluorocarbons
Three Bond Technical News Issued Dec. 20, 1989 An Overview of Fluorocarbons Introduction Currently, chlorofluorocarbons are garnering a lot of Three Bond Co,. Ltd. has been working night and day in attention around the world. Chlorofluorocarbons are used in search of alternatives to chlorofluorocarbons. In the future, many of our products and when we look at the future of products that do not contain any chlorofluorocarbons will chlorofluorocarbons in the world, and in Japan, we can replace products that now contain chlorofluorocarbons. To foresee that their use will be strictly limited or eliminated all ensure these products reach our customers as smoothly as together. possible, it is important that they have a good understanding of the issues surrounding chlorofluorocarbons. Whatever the case, Three Bond Co., Ltd. is obligated to provide our customers with products that have the same functionality as chlorofluorocarbons. Contents Introduction ................................................................................................................................................................1 1. What is a fluorocarbon? .......................................................................................................................................2 2. Properties of fluorocarbons ..................................................................................................................................2 3. Types of fluorocarbons.........................................................................................................................................2 -

Halocarbon 41, HFC-41
SAFETY DATA SHEET Fluoromethane Issue Date: 16.01.2013 Version: 1.1 SDS No.: 000010021722 Last revised date: 19.03.2021 1/26 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifier Product name : Fluoromethane Other Name : R41, Halocarbon 41, HFC-41 Additional identification Chemical name: Fluoromethane Chemical formula: CH3F INDEX No. - CAS-No. 593-53-3 EC No. 209-796-6 REACH Registration No. 01-2120746661-53 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Industrial and professional. Perform risk assessment prior to use. Aerosol propellant. Use as an Intermediate (transported, on-site isolated). Use for electronic component manufacture. Using gas alone or in mixtures for the calibration of analysis equipment. Uses advised against Consumer use. 1.3 Details of the supplier of the safety data sheet Supplier BOC Telephone : 0800 111 333 Priestley Road, Worsley M28 2UT Manchester E-mail: [email protected] 1.4 Emergency telephone number: 0800 111 333 SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 as amended. Physical Hazards Gases under pressure Liquefied gas H280: Contains gas under pressure; may explode if heated. Flammable gas Category 1 H220: Extremely flammable gas. SDS_GB - 000010021722 SAFETY DATA SHEET Fluoromethane Issue Date: 16.01.2013 Version: 1.1 SDS No.: 000010021722 Last revised date: 19.03.2021 2/26 2.2 Label Elements Signal Word : Danger Hazard Statement(s) : H220: Extremely flammable gas. H280: Contains gas under pressure; may explode if heated. -

Refrigerants
Related Commercial Resources CHAPTER 29 REFRIGERANTS Refrigerant Properties .................................................................................................................. 29.1 Refrigerant Performance .............................................................................................................. 29.6 Safety ............................................................................................................................................. 29.6 Leak Detection .............................................................................................................................. 29.6 Effect on Construction Materials .................................................................................................. 29.9 EFRIGERANTS are the working fluids in refrigeration, air- cataracts, and impaired immune systems. It also can damage sensi- R conditioning, and heat-pumping systems. They absorb heat tive crops, reduce crop yields, and stress marine phytoplankton (and from one area, such as an air-conditioned space, and reject it into thus human food supplies from the oceans). In addition, exposure to another, such as outdoors, usually through evaporation and conden- UV radiation degrades plastics and wood. sation, respectively. These phase changes occur both in absorption Stratospheric ozone depletion has been linked to the presence of and mechanical vapor compression systems, but not in systems oper- chlorine and bromine in the stratosphere. Chemicals with long ating on a gas cycle using a -

DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS
criteria for a recommended standard . occupational exposure to DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE criteria for a recommended standard... OCCUPATIONAL EXPOSURE TO DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE Public Health Service Center for Disease Control National Institute for Occupational Safety and Health September 1977 For sale by the Superintendent of Documents, U.S. Government Printing Office, Washington, D.C. 20402 DHEW (NIOSH) Publication No. 77-193 PREFACE The Occupational Safety and Health Act of 1970 emphasizes the need for standards to protect the health and safety of workers exposed to an ever-increasing number of potential hazards at their workplace. The National Institute for Occupational Safety and Health has projected a formal system of research, with priorities determined on the basis of specified indices, to provide relevant data from which valid criteria for effective standards can be derived. Recommended standards for occupational exposure, which are the result of this work, are based on the health effects of exposure. The Secretary of Labor will weigh these recommendations along with other considerations such as feasibility and means of implementation in developing regulatory standards. It is intended to present successive reports as research and epidemiologic studies are completed and as sampling and analytical methods are developed. Criteria and standards will be reviewed periodically to ensure continuing protection of the worker. I am pleased to acknowledge the contributions to this report on the decomposition products of fluorocarbon polymers by members of the NIOSH staff and the valuable constructive comments by the Review Consultants on the Decomposition Products of Fluorocarbon Polymers and by Robert B. -

Fluoromethane (R41) 059
Page : 1 / 9 SAFETY DATA SHEET Revised edition no : 3 - 00 BC in accordance with REACH Date : 31 / 1 / 2014 regulation 1907/2006/EC Supersedes : 2 / 10 / 2012 Fluoromethane (R41) 059 SECTION 1. Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Trade name : Fluoromethane (R41) SDS Nr : 059 Chemical description : Fluoromethane (R41) CAS No :593-53-3 EC No :209-796-6 Index No :--- Registration-No. : Registration deadline not expired. Chemical formula : CH3F 1.2. Relevant identified uses of the substance or mixture and uses advised against Relevant identified uses : Test gas / Calibration gas. Industrial and professional. Perform risk assessment prior to use. Laboratory use. Contact supplier for more uses information. Use as refrigerant. Chemical reaction / Synthesis. 1.3. Details of the supplier of the safety data sheet Company identification : AIR LIQUIDE Deutschland GmbH Hans-Günther-Sohl-Straße 5 D-40235 Düsseldorf GERMANY Telefon: +49 (0)211 6699-0 - Fax: +49 (0)211 6699-222 E-Mail address (competent person) : [email protected] 1.4. Emergency telephone number Emergency telephone number : +49 (0)2151 398668 SECTION 2. Hazards identification 2.1. Classification of the substance or mixture Hazard Class and Category Code(s), Regulation (EC) No 1272/2008 (CLP) • Physical hazards : Flammable gases - Category 1 - Danger - (CLP : Flam. Gas 1) - H220 Gases under pressure - Liquefied gas - Warning - (CLP : Press. Gas) - H280 Classification EC 67/548 or EC 1999/45 Classification : F+; R12 Not included in Annex VI. 2.2. Label elements Labelling Regulation EC 1272/2008 (CLP) • Hazard pictograms M— M« • Hazard pictograms code : GHS02 - GHS04 • Signal words : Danger • Hazard statements : H220 - Extremely flammable gas. -

An Electron Impact Investigation of the Fluorinated Methanes
AN ELECTRON IMPACT INVESTIGATION OF THE FLUORINATED METHANES DISSERTATION Presented In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of the Ohio State University By EDWIN HENRY LOUGHER, B.S. The Ohio State University 1952 Approved by Advisor ACKNOWLEDGEMENT I wish to express my appreciation to Dr. E. N. Lassettre for his assistance during the course of this investigation. His interest and grasp of the problems involved were a principal factor in the completion of the work. I should like to thank Dr. A. L. Henne for valuable advice and for some of the materials used in this investigation. I wish to thank Dr. A. B. Garrett, the Chemistry Department, and the Ohio State Research Foundation for the assistantships which enabled me to carry this work to completion. Finally, I wish to thank my wife for her continued encouragement and for typing this dissertation. 8 0 0 4 6 1 TABLE OF CONTENTS Page Introduction 1 I. History and Theory 2 A. History of Electron Impact Work 2 B. General Theory of Electron Impact Spectra 3 C. Localization of Valence Electrons 8 (1) The Concept of Localization 8 (2) Electron Impact Spectra of Saturated and Unsaturated Hydrocarbons 10 (3) Localized and Non Localized Molecular Orbitals 13 (4) Localized and Non Localized Molecular Orbitals in Excitation Processes 21 D. Choice of Compounds 23 II. The Electron Spectrometer 24 A. General Description 24 B. Modification of the Apparatus 32 C. Operational Procedure 35 III. Experimental Work 39 A. Materials 39 B. Graphs of Observed Spectra 43 C. -

SAFETY DATASHEET Revision Date : 06/2021
Page : 1/10 Revised edition n° : 10.1 SAFETY DATASHEET Revision date : 06/2021 MTG059 Fluoromethane (R41) SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Trade name Fluoromethane (R41) Chemical description Fluoromethane, Methyl fluoride CAS N° 593-53-3 CE N° 209-796-6 Index N° -- Registration n° 01-2120746661-53 Chemical formula CH3F 1.2. Relevant identified uses of the substance or mixture and uses advised against Relevant identified uses Industrial and professional Test gas/Calibration gas Chemical reaction / Synthesis Use as refrigerant Laboratory use Polymer production. Contact supplier for more information on uses Uses advised against Consumer use not recommended 1.3. Details of the supplier of the safety data sheet MULTIGAS Company identification Route de l’Industrie 102 CH-1564 Domdidier Phone number +41 (0) 26 676 94 94 E-mail address [email protected] 1.4. Emergency telephone numbers 145 (Toxicology Centre Zurich) or +41 (0) 44 251 51 51 +41 (0) 26 676 94 94 (Multigas) SECTION 2: Hazards identification 2.1. Classification of the substance or mixture Classification according to Regulation (EC) No. 1272/2008 [CLP] Physical hazards Flammable gases, Category 1 H220 Gases under pressure : Liquefied gas H280 Page : 2/10 Revised edition n° : 10.1 SAFETY DATASHEET Revision date : 06/2021 MTG059 Fluoromethane (R41) For the complete H-sentences texts mentioned in that chapter, refer to Section 16 2.2. Label elements Labelling according to Regulation (EC) No. 1272/2008 [CLP] Hazard pictograms GHS02 GHS04 Signal word Danger Hazard statements H220 Extremely flammable gas H280 Contains gas under pressure; may explode if heated Precautionary statements P210 Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. -

C1 and Cz FLUORINATED HYDROCARBON EFFECTS on the EXTINCTION CHARACTERISTICS of METHANE VS
Presented at the Halon Opiions Technical Working Conference, Albuquerque, NM,May 6-8, 1997 C1 AND Cz FLUORINATED HYDROCARBON EFFECTS ON THE EXTINCTION CHARACTERISTICS OF METHANE VS. AIR COUNTERFLOW DIFFUSION FLAMES Michael A. Tanoff', Richard R. Dobbins, Mitchell D. Smooke Department of Mechanical Engineering, Yale University P.O. Boz 208284 New Haven, Connecticut, U.S.A. 06520-8284 Donald R. Burgess, Jr., Michael R. Zachariah, Wing Tsang Chemical Science and Technology Laboratory National Institute of Standanls and Technology Gaithersburg, Maryland, U.S.A. 20899-0001 Phillip R. Westmoreland Department of Chemical Engineering, University of Massachusetts Amherst, Massachusetts, U.S.A. 01009-9110 ABSTRACT Detailed numerical calculations are compared against available experiments and are used to inveg tigate the flame suppression and extinction properties of a variety of flourinated hydrocarbon candidates selected for halon 1301 and halon 1211 replacement. The air stream in a methane vs. air counterflow diffu- sion flame is doped with varying amountsof ChF,CH2F2, CHF3, CF4, CHFz-CFs and CHzF-CF3, as well as the diluents N2 and COz, and their effect on the strain rate required for flame extinction is monitored. The chemical model is based on a recently developed set of thermochemical and chemical kinetic data for the C1 and Cz fluorinated hydrocarbons. Numerical results are in very good agreement with available experimental data for CF4- and CHF3- suppresed methane vs. air counterflow diffusion flames. Both computations and experiments show similar linear decreases in required extinction strain rate with molar per cent agent added to the oxidizer stream, thereby yielding the same effectiveness for suppressing flames.