Electronic Supplementary Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Genomic Evidence of Reactive Oxygen Species Elevation in Papillary Thyroid Carcinoma with Hashimoto Thyroiditis
Endocrine Journal 2015, 62 (10), 857-877 Original Genomic evidence of reactive oxygen species elevation in papillary thyroid carcinoma with Hashimoto thyroiditis Jin Wook Yi1), 2), Ji Yeon Park1), Ji-Youn Sung1), 3), Sang Hyuk Kwak1), 4), Jihan Yu1), 5), Ji Hyun Chang1), 6), Jo-Heon Kim1), 7), Sang Yun Ha1), 8), Eun Kyung Paik1), 9), Woo Seung Lee1), Su-Jin Kim2), Kyu Eun Lee2)* and Ju Han Kim1)* 1) Division of Biomedical Informatics, Seoul National University College of Medicine, Seoul, Korea 2) Department of Surgery, Seoul National University Hospital and College of Medicine, Seoul, Korea 3) Department of Pathology, Kyung Hee University Hospital, Kyung Hee University School of Medicine, Seoul, Korea 4) Kwak Clinic, Okcheon-gun, Chungbuk, Korea 5) Department of Internal Medicine, Uijeongbu St. Mary’s Hospital, Uijeongbu, Korea 6) Department of Radiation Oncology, Seoul St. Mary’s Hospital, Seoul, Korea 7) Department of Pathology, Chonnam National University Hospital, Kwang-Ju, Korea 8) Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea 9) Department of Radiation Oncology, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Seoul, Korea Abstract. Elevated levels of reactive oxygen species (ROS) have been proposed as a risk factor for the development of papillary thyroid carcinoma (PTC) in patients with Hashimoto thyroiditis (HT). However, it has yet to be proven that the total levels of ROS are sufficiently increased to contribute to carcinogenesis. We hypothesized that if the ROS levels were increased in HT, ROS-related genes would also be differently expressed in PTC with HT. To find differentially expressed genes (DEGs) we analyzed data from the Cancer Genomic Atlas, gene expression data from RNA sequencing: 33 from normal thyroid tissue, 232 from PTC without HT, and 60 from PTC with HT. -

European Patent Office U.S. Patent and Trademark Office
EUROPEAN PATENT OFFICE U.S. PATENT AND TRADEMARK OFFICE CPC NOTICE OF CHANGES 89 DATE: JULY 1, 2015 PROJECT RP0098 The following classification changes will be effected by this Notice of Changes: Action Subclass Group(s) Symbols deleted: C12Y 101/01063 C12Y 101/01128 C12Y 101/01161 C12Y 102/0104 C12Y 102/03011 C12Y 103/01004 C12Y 103/0103 C12Y 103/01052 C12Y 103/99007 C12Y 103/9901 C12Y 103/99013 C12Y 103/99021 C12Y 105/99001 C12Y 105/99002 C12Y 113/11013 C12Y 113/12012 C12Y 114/15002 C12Y 114/99028 C12Y 204/01119 C12Y 402/01052 C12Y 402/01058 C12Y 402/0106 C12Y 402/01061 C12Y 601/01025 C12Y 603/02027 Symbols newly created: C12Y 101/01318 C12Y 101/01319 C12Y 101/0132 C12Y 101/01321 C12Y 101/01322 C12Y 101/01323 C12Y 101/01324 C12Y 101/01325 C12Y 101/01326 C12Y 101/01327 C12Y 101/01328 C12Y 101/01329 C12Y 101/0133 C12Y 101/01331 C12Y 101/01332 C12Y 101/01333 CPC Form – v.4 CPC NOTICE OF CHANGES 89 DATE: JULY 1, 2015 PROJECT RP0098 Action Subclass Group(s) C12Y 101/01334 C12Y 101/01335 C12Y 101/01336 C12Y 101/01337 C12Y 101/01338 C12Y 101/01339 C12Y 101/0134 C12Y 101/01341 C12Y 101/01342 C12Y 101/03043 C12Y 101/03044 C12Y 101/98003 C12Y 101/99038 C12Y 102/01083 C12Y 102/01084 C12Y 102/01085 C12Y 102/01086 C12Y 103/01092 C12Y 103/01093 C12Y 103/01094 C12Y 103/01095 C12Y 103/01096 C12Y 103/01097 C12Y 103/0701 C12Y 103/08003 C12Y 103/08004 C12Y 103/08005 C12Y 103/08006 C12Y 103/08007 C12Y 103/08008 C12Y 103/08009 C12Y 103/99032 C12Y 104/01023 C12Y 104/01024 C12Y 104/03024 C12Y 105/01043 C12Y 105/01044 C12Y 105/01045 C12Y 105/03019 C12Y 105/0302 -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -
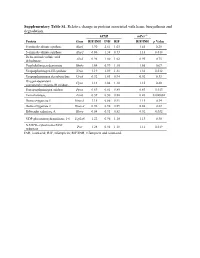
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

(12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur Et Al
US 2012026.6329A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur et al. (43) Pub. Date: Oct. 18, 2012 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/10 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/24 (2006.01) CI2N 9/02 (2006.01) (75) Inventors: Eric J. Mathur, Carlsbad, CA CI2N 9/06 (2006.01) (US); Cathy Chang, San Marcos, CI2P 2L/02 (2006.01) CA (US) CI2O I/04 (2006.01) CI2N 9/96 (2006.01) (73) Assignee: BP Corporation North America CI2N 5/82 (2006.01) Inc., Houston, TX (US) CI2N 15/53 (2006.01) CI2N IS/54 (2006.01) CI2N 15/57 2006.O1 (22) Filed: Feb. 20, 2012 CI2N IS/60 308: Related U.S. Application Data EN f :08: (62) Division of application No. 1 1/817,403, filed on May AOIH 5/00 (2006.01) 7, 2008, now Pat. No. 8,119,385, filed as application AOIH 5/10 (2006.01) No. PCT/US2006/007642 on Mar. 3, 2006. C07K I4/00 (2006.01) CI2N IS/II (2006.01) (60) Provisional application No. 60/658,984, filed on Mar. AOIH I/06 (2006.01) 4, 2005. CI2N 15/63 (2006.01) Publication Classification (52) U.S. Cl. ................... 800/293; 435/320.1; 435/252.3: 435/325; 435/254.11: 435/254.2:435/348; (51) Int. Cl. 435/419; 435/195; 435/196; 435/198: 435/233; CI2N 15/52 (2006.01) 435/201:435/232; 435/208; 435/227; 435/193; CI2N 15/85 (2006.01) 435/200; 435/189: 435/191: 435/69.1; 435/34; CI2N 5/86 (2006.01) 435/188:536/23.2; 435/468; 800/298; 800/320; CI2N 15/867 (2006.01) 800/317.2: 800/317.4: 800/320.3: 800/306; CI2N 5/864 (2006.01) 800/312 800/320.2: 800/317.3; 800/322; CI2N 5/8 (2006.01) 800/320.1; 530/350, 536/23.1: 800/278; 800/294 CI2N I/2 (2006.01) CI2N 5/10 (2006.01) (57) ABSTRACT CI2N L/15 (2006.01) CI2N I/19 (2006.01) The invention provides polypeptides, including enzymes, CI2N 9/14 (2006.01) structural proteins and binding proteins, polynucleotides CI2N 9/16 (2006.01) encoding these polypeptides, and methods of making and CI2N 9/20 (2006.01) using these polynucleotides and polypeptides. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -

Nitrite Reduction by Xanthine Oxidase Family Enzymes: a New Class of Nitrite Reductases
J Biol Inorg Chem (2011) 16:443–460 DOI 10.1007/s00775-010-0741-z ORIGINAL PAPER Nitrite reduction by xanthine oxidase family enzymes: a new class of nitrite reductases Luisa B. Maia • Jose´ J. G. Moura Received: 9 August 2010 / Accepted: 19 November 2010 / Published online: 19 December 2010 Ó SBIC 2010 Abstract Mammalian xanthine oxidase (XO) and Des- Keywords Nitrite reduction Á Nitric oxide formation Á ulfovibrio gigas aldehyde oxidoreductase (AOR) are Molybdenum Á Xanthine oxidase Á Aldehyde members of the XO family of mononuclear molybdoen- oxidoreductase zymes that catalyse the oxidative hydroxylation of a wide range of aldehydes and heterocyclic compounds. Much less Abbreviations known is the XO ability to catalyse the nitrite reduction to AOR Aldehyde oxidoreductase nitric oxide radical (NO). To assess the competence of other DMSOR Dimethylsulfoxide reductase XO family enzymes to catalyse the nitrite reduction and to EPR Electron paramagnetic resonance shed some light onto the molecular mechanism of this Fe/S Iron–sulfur centre reaction, we characterised the anaerobic XO- and AOR- Fe/S–NO Dinitrosyl–iron–sulfur complex catalysed nitrite reduction. The identification of NO as the (MGD) –Fe Ferrous complex of di(N-methyl-D- reaction product was done with a NO-selective electrode 2 glucamine dithiocarbamate) and by electron paramagnetic resonance (EPR) spectros- (MGD) –Fe–NO Mononitrosyl–iron complex copy. The steady-state kinetic characterisation corroborated 2 Mo-enzymes Pterin–molybdenum-containing the XO-catalysed nitrite reduction and demonstrated, for the enzymes first time, that the prokaryotic AOR does catalyse the nitrite NaR Nitrate reductases reduction to NO, in the presence of any electron donor to the NO Nitric oxide radical enzyme, substrate (aldehyde) or not (dithionite). -
Generated by SRI International Pathway Tools Version 25.0, Authors S
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000411195-HmpCyc: Delftia acidovorans CCUG 274B Cellular Overview Connections between pathways are omitted for legibility. sn-glycerol a dicarboxylate a dicarboxylate an amino an amino an amino an amino an amino an amino an amino 3-phosphate O-acetyl-L-serine an amino an amino acid acid acid acid acid acid acid an aromatic an amino an amino an amino an amino an amino an amino an amino an amino an amino an amino cys sn-glycerol acid acid glutamate glutamate glutamate Fe 2+ amino acid acid acid acid acid acid acid predicted 3-phosphate acid acid acid acid enterobactin ABC predicted predicted predicted predicted predicted predicted predicted transporter ABC ABC ABC ABC ABC ABC ABC RS32735 RS04550 RS10535 RS13605 RS15475 RS15470 RS16910 RS30270 RS11330 RS26660 RS26655 RS18485 RS05155 RS21560 RS30425 RS15600 RS05020 RS11425 RS02945 RS07585 RS30175 RS02895 RS07555 RS09960 RS17430 RS02730 RS10335 RS10680 RS16050 RS22970 RS06675 RS08770 RS30075 RS04655 RS08420 RS10940 RS25135 RS22045 RS29235 RS08015 RS04605 RS08300 RS10155 RS28975 RS13905 RS13355 RS04600 RS03585 RS04625 RS21235 RS04635 RS08415