Mechanisms of Autoreceptor-Mediated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The B Vitamins Nicotinamide (B3) and Riboflavin (B2)
The B Vitamins Nicotinamide (B3) and Riboflavin (B2) Stimulate Metamorphosis in Larvae of the Deposit- Feeding Polychaete Capitella teleta: Implications for a Sensory Ligand-Gated Ion Channel Robert T. Burns1*, Jan A. Pechenik1, William J. Biggers2, Gia Scavo2, Christopher Lehman2 1 Department of Biology, Tufts University, Medford, Massachusetts, United States of America, 2 Department of Biology, Wilkes University, Wilkes-Barre, Pennsylvania, United States of America Abstract Marine sediments can contain B vitamins, presumably incorporated from settled, decaying phytoplankton and microorganisms associated with decomposition. Because B vitamins may be advantageous for the energetically intensive processes of metamorphosis, post-metamorphic growth, and reproduction, we tested several B vitamins to determine if they would stimulate larvae of the deposit-feeding polychaete Capitella teleta to settle and metamorphose. Nicotinamide and riboflavin individually stimulated larvae of C. teleta to settle and metamorphose, generally within 1–2 hours at nicotinamide concentrations as low as 3 mM and riboflavin concentrations as low as 50 mM. More than 80% of the larvae metamorphosed within 30 minutes at a nicotinamide concentration of 7 mM. The pyridine channel agonist pyrazinecarboxamide also stimulated metamorphosis at very low concentrations. In contrast, neither lumichrome, thiamine HCl, pyridoxine HCl, nor vitamin B12 stimulated larvae of C. teleta to metamorphose at concentrations as high as 500 mM. Larvae also did not metamorphose in response to either nicotinamide or pyrazinecarboxamide in calcium-free seawater or with the addition of 4-acetylpyridine, a competitive inhibitor of the pyridine receptor. Together, these results suggest that larvae of C. teleta are responding to nicotinamide and riboflavin via a chemosensory pyridine receptor similar to that previously reported to be present on crayfish chela and involved with food recognition. -

Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment Via CREB-BDNF Signalling
International Journal of Molecular Sciences Article Neural Stem Cell-Derived Exosomes Revert HFD-Dependent Memory Impairment via CREB-BDNF Signalling Matteo Spinelli 1, Francesca Natale 1,2, Marco Rinaudo 1, Lucia Leone 1,2, Daniele Mezzogori 1, Salvatore Fusco 1,2,* and Claudio Grassi 1,2 1 Department of Neuroscience, Università Cattolica del Sacro Cuore, 00168 Rome, Italy; [email protected] (M.S.); [email protected] (F.N.); [email protected] (M.R.); [email protected] (L.L.); [email protected] (D.M.); [email protected] (C.G.) 2 Fondazione Policlinico Universitario Agostino Gemelli IRCCS, 00168 Rome, Italy * Correspondence: [email protected] Received: 6 October 2020; Accepted: 25 November 2020; Published: 26 November 2020 Abstract: Overnutrition and metabolic disorders impair cognitive functions through molecular mechanisms still poorly understood. In mice fed with a high fat diet (HFD) we analysed the expression of synaptic plasticity-related genes and the activation of cAMP response element-binding protein (CREB)-brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) signalling. We found that a HFD inhibited both CREB phosphorylation and the expression of a set of CREB target genes in the hippocampus. The intranasal administration of neural stem cell (NSC)-derived exosomes (exo-NSC) epigenetically restored the transcription of Bdnf, nNOS, Sirt1, Egr3, and RelA genes by inducing the recruitment of CREB on their regulatory sequences. Finally, exo-NSC administration rescued both BDNF signalling and memory in HFD mice. Collectively, our findings highlight novel mechanisms underlying HFD-related memory impairment and provide evidence of the potential therapeutic effect of exo-NSC against metabolic disease-related cognitive decline. -

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

Lgr5 Homologues Associate with Wnt Receptors and Mediate R-Spondin Signalling
ARTICLE doi:10.1038/nature10337 Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling Wim de Lau1*, Nick Barker1{*, Teck Y. Low2, Bon-Kyoung Koo1, Vivian S. W. Li1, Hans Teunissen1, Pekka Kujala3, Andrea Haegebarth1{, Peter J. Peters3, Marc van de Wetering1, Daniel E. Stange1, Johan E. van Es1, Daniele Guardavaccaro1, Richard B. M. Schasfoort4, Yasuaki Mohri5, Katsuhiko Nishimori5, Shabaz Mohammed2, Albert J. R. Heck2 & Hans Clevers1 The adult stem cell marker Lgr5 and its relative Lgr4 are often co-expressed in Wnt-driven proliferative compartments. We find that conditional deletion of both genes in the mouse gut impairs Wnt target gene expression and results in the rapid demise of intestinal crypts, thus phenocopying Wnt pathway inhibition. Mass spectrometry demonstrates that Lgr4 and Lgr5 associate with the Frizzled/Lrp Wnt receptor complex. Each of the four R-spondins, secreted Wnt pathway agonists, can bind to Lgr4, -5 and -6. In HEK293 cells, RSPO1 enhances canonical WNT signals initiated by WNT3A. Removal of LGR4 does not affect WNT3A signalling, but abrogates the RSPO1-mediated signal enhancement, a phenomenon rescued by re-expression of LGR4, -5 or -6. Genetic deletion of Lgr4/5 in mouse intestinal crypt cultures phenocopies withdrawal of Rspo1 and can be rescued by Wnt pathway activation. Lgr5 homologues are facultative Wnt receptor components that mediate Wnt signal enhancement by soluble R-spondin proteins. These results will guide future studies towards the application of R-spondins for regenerative purposes of tissues expressing Lgr5 homologues. The genes Lgr4, Lgr5 and Lgr6 encode orphan 7-transmembrane 4–5 post-induction onwards. -

Profiling G Protein-Coupled Receptors of Fasciola Hepatica Identifies Orphan Rhodopsins Unique to Phylum Platyhelminthes
bioRxiv preprint doi: https://doi.org/10.1101/207316; this version posted October 23, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Profiling G protein-coupled receptors of Fasciola hepatica 2 identifies orphan rhodopsins unique to phylum 3 Platyhelminthes 4 5 Short title: Profiling G protein-coupled receptors (GPCRs) in Fasciola hepatica 6 7 Paul McVeigh1*, Erin McCammick1, Paul McCusker1, Duncan Wells1, Jane 8 Hodgkinson2, Steve Paterson3, Angela Mousley1, Nikki J. Marks1, Aaron G. Maule1 9 10 11 1Parasitology & Pathogen Biology, The Institute for Global Food Security, School of 12 Biological Sciences, Queen’s University Belfast, Medical Biology Centre, 97 Lisburn 13 Road, Belfast, BT9 7BL, UK 14 15 2 Institute of Infection and Global Health, University of Liverpool, Liverpool, UK 16 17 3 Institute of Integrative Biology, University of Liverpool, Liverpool, UK 18 19 * Corresponding author 20 Email: [email protected] 21 1 bioRxiv preprint doi: https://doi.org/10.1101/207316; this version posted October 23, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 22 Abstract 23 G protein-coupled receptors (GPCRs) are established drug targets. Despite their 24 considerable appeal as targets for next-generation anthelmintics, poor understanding 25 of their diversity and function in parasitic helminths has thwarted progress towards 26 GPCR-targeted anti-parasite drugs. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

The Role of the Serotonergic System and the Effects of Antidepressants During Brain Development Examined Using in Vivo Pet Imaging and in Vitro Receptor Binding
From THE DEPARTMENT OF CLINICAL NEUROSCIENCE Karolinska Institutet, Stockholm, Sweden THE ROLE OF THE SEROTONERGIC SYSTEM AND THE EFFECTS OF ANTIDEPRESSANTS DURING BRAIN DEVELOPMENT EXAMINED USING IN VIVO PET IMAGING AND IN VITRO RECEPTOR BINDING Stal Saurav Shrestha Stockholm 2014 Cover Illustration: Voxel-wise analysis of the whole monkey brain using the PET radioligand, [11C]DASB showing persistent serotonin transporter upregulation even after more than 1.5 years of fluoxetine discontinuation. All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Universitetsservice-AB © Stal Saurav Shrestha, 2014 ISBN 978-91-7549-522-4 Serotonergic System and Antidepressants During Brain Development To my family Amaze yourself ! Stal Saurav Shrestha, 2014 The Department of Clinical Neuroscience The role of the serotonergic system and the effects of antidepressants during brain development examined using in vivo PET imaging and in vitro receptor binding AKADEMISK AVHANDLING som för avläggande av medicine doktorsexamen vid Karolinska Institutet offentligen försvaras i CMM föreläsningssalen L8:00, Karolinska Universitetssjukhuset, Solna THESIS FOR DOCTORAL DEGREE (PhD) Stal Saurav Shrestha Date: March 31, 2014 (Monday); Time: 10 AM Venue: Center for Molecular Medicine Lecture Hall Floor 1, Karolinska Hospital, Solna Principal Supervisor: Opponent: Robert B. Innis, MD, PhD Klaus-Peter Lesch, MD, PhD National Institutes of Health University of Würzburg Department of NIMH Department -

Understanding Allergic Multimorbidity Within the Non-Eosinophilic
University of Groningen Understanding allergic multimorbidity within the non-eosinophilic interactome Aguilar, Daniel; Lemonnier, Nathanael; Koppelman, Gerard H; Melén, Erik; Oliva, Baldo; Pinart, Mariona; Guerra, Stefano; Bousquet, Jean; Anto, Josep M Published in: PLoS ONE DOI: 10.1371/journal.pone.0224448 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2019 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Aguilar, D., Lemonnier, N., Koppelman, G. H., Melén, E., Oliva, B., Pinart, M., Guerra, S., Bousquet, J., & Anto, J. M. (2019). Understanding allergic multimorbidity within the non-eosinophilic interactome. PLoS ONE, 14(11), [e0224448]. https://doi.org/10.1371/journal.pone.0224448 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 26-12-2020 RESEARCH ARTICLE Understanding allergic multimorbidity within the non-eosinophilic interactome 1,2,3 4 5,6 7 Daniel AguilarID *, Nathanael LemonnierID , Gerard H. -

Exploring the Heteromeric Interface of the 5-HT2A-Mglu2 Receptor Complex
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2020 Exploring the Heteromeric Interface of the 5-HT2A-mGlu2 Receptor Complex Mohamed Aarif Abdul Kareem Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Nervous System Diseases Commons, Other Psychiatry and Psychology Commons, Physiological Processes Commons, and the Translational Medical Research Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/6235 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Exploring the Heteromeric Interface of the 5-HT2A-mGlu2 Receptor Complex A Thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Physiology and Biophysics at Virginia Commonwealth University By: Mohamed Aarif Abdul Kareem B.A. Neurobiology, Boston University, 2018 Mentor: Javier González-Maeso Associate Professor Department of Physiology and Biophysics Virginia Commonwealth University Richmond, Virginia April 30, 2020 Acknowledgments: Thank you to my peers for their continued support of my dreams and aspirations. Thank you to my mentors for pushing and supporting me every step of the way. Thank you to Virginia Commonwealth University for providing opportunities which foster my passion for science and allow it to continue to flourish. Indeed, I owe much gratitude to my parents, Abdul & Jasmine Kareem, who have guided me in becoming a strong, independent student, and encourage me to take calculated risks and face challenges head on. -
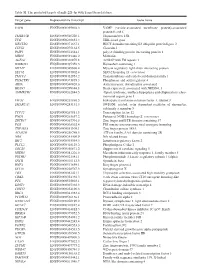
Table SI. the Predicted Targets of Mir-221-3P with Targetscan Database
Table SI. The predicted targets of miR-221-3p with TargetScan database. Target gene Representative transcript Gene name VAPB ENST00000395802.3 VAMP (vesicle-associated membrane protein)-associated protein B and C TMSB15B ENST00000540220.1 Thymosin beta 15B TFG ENST00000240851.4 TRK-fused gene HECTD2 ENST00000371667.1 HECT domain containing E3 ubiquitin protein ligase 2 CLVS2 ENST00000275162.5 Clavesin 2 PAIP1 ENST00000514514.1 poly(A) binding protein interacting protein 1 MIDN ENST00000591446.2 Midnolin AGFG1 ENST00000310078.8 ArfGAP with FG repeats 1 HMBOX1 ENST00000397358.3 Homeobox containing 1 MYLIP ENST00000349606.4 Myosin regulatory light chain interacting protein SEC62 ENST00000337002.4 SEC62 homolog (S. cerevisiae) TMCC1 ENST00000432054.2 Transmembrane and coiled-coil domain family 1 PHACTR4 ENST00000373839.3 Phosphatase and actin regulator 4 AIDA ENST00000340020.6 Axin interactor, dorsalization associated BEAN1 ENST00000299694.8 Brain expressed, associated with NEDD4, 1 AMMECR1 ENST00000262844.5 Alport syndrome, midface hypoplasia and elliptocytosis chro- mosomal region gene 1 EIF3J ENST00000261868.5 Eukaryotic translation initiation factor 3, subunit J SMARCA5 ENST00000283131.3 SWI/SNF related, actin dependent regulator of chromatin, subfamily a, member 5 TCF12 ENST00000267811.5 Transcription factor 12 PNO1 ENST00000263657.2 Partner of NOB1 homolog (S. cerevisiae) ZBTB37 ENST00000367701.5 Zinc finger and BTB domain containing 37 FOS ENST00000303562.4 FBJ murine osteosarcoma viral oncogene homolog ZNF385A ENST00000551109.1 Zinc -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. The CX3CR1/CX3CL1 Axis Drives the Migration and Maturation of Oligodendroglia in the Central Nervous System Catriona Ford Thesis Submitted for the Degree of Doctor of Philosophy The University of Edinburgh 2017 Abstract In the central nervous system, the axons of neurons are protected from damage and aided in electrical conductivity by the myelin sheath, a complex of proteins and lipids formed by oligodendrocytes. Loss or damage to the myelin sheath may result in impairment of electrical axonal conduction and eventually to neuronal death. Such demyelination is responsible, at least in part, for the disabling neurodegeneration observed in pathologies such as Multiple Sclerosis (MS) and Spinal Cord Injury. In the regenerative process of remyelination, oligodendrocyte precursor cells (OPCs), the resident glial stem cell population of the adult CNS, migrate toward the injury site, proliferate and differentiate into adult oligodendrocytes which subsequently reform the myelin sheath. -

Functional Analysis of the Latrophilin Homolog Dcirl in Drosophila Melanogaster
Functional analysis of the latrophilin homolog dCirl in Drosophila melanogaster Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Julius-Maximilians-Universität Würzburg vorgelegt von Jennifer Gehring aus Würzburg Physiologisches Institut – Lehrstuhl für Physiologie Schwerpunkt Neurophysiologie Würzburg, 2014 Eingereicht am: ………………………… Mitglieder der Promotionskommission: Vorsitzender: ………………………… Gutachter: Prof. Dr. Manfred Heckmann Gutachter: Prof. Dr. Christian Stigloher Tag des Promotionskolloquiums: ………………………... Doktorurkunde ausgehändigt am: .................................. I Erklärung Ich versichere hiermit, dass ich die vorliegende Dissertation mit dem Titel “Functional analysis of the latrophilin homolog dCirl in Drosophila melanogaster” eigenständig angefertigt und mich dabei keiner anderen als der von mir angegebenen Quellen und Hilfsmittel bedient habe. Die Dissertation wurde in der jetzigen oder einer ähnlichen Form noch bei keiner anderen Hochschule eingereicht. Würzburg, 2014 ____________________________ Jennifer Gehring II Contents Contents 1. Summary............... ................................................................................................. 1 2. Introduction........... .................................................................................................. 4 2.1 Adhesion class G-protein coupled receptors ..................................................... 4 2.1.1 Latrophilin – a prototype of adhesion-GPCRs ............................................ 6 2.1.2 Structural