Tutorial Review: X-Ray Mapping in Electron-Beam Instruments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Electron Microprobe Analysis Course 12.141 Notes MIT Electron
Electron Microprobe Analysis Course 12.141 Notes Dr. Nilanjan Chatterjee The electron microprobe provides a complete micron-scale quantitative chemical analysis of inorganic solids. The method is nondestructive and utilizes characteristic x-rays excited by an electron beam incident on a flat surface of the sample. This course provides an introduction to the theory of x-ray microanalysis by wavelength and energy dispersive spectrometry (WDS and EDS), ZAF matrix correction procedures, and scanning electron imaging techniques using backscattered electron (BE), secondary electron (SE), x-ray (elemental mapping), and cathodoluminescence (CL) signals. Lab sessions involve hands- on use of the JEOL JXA-8200 Superprobe. MIT Electron Microprobe Facility Massachusetts Institute of Technology Department of Earth, Atmospheric & Planetary Sciences Room: 54-1221, Cambridge, MA 02139 Phone: (617) 253-1995; Fax: (617) 253-7102 e-mail: [email protected] web: http://web.mit.edu/e-probe/www/ Nilanjan ChatterjeeMIT, Cambridge, MA, USA October 2017 2 TABLE OF CONTENTS Page number 1. INTRODUCTION 3 2. ELECTRON SPECIMEN INTERACTIONS 5 2.1. ELASTIC SCATTERING 5 2.1.1. Electron backscattering 5 2.1.2. Electron interaction volume 6 2.2. INELASTIC SCATTERING 7 2.2.1. Secondary electron generation 7 2.2.2. Characteristic x-ray generation: inner-shell ionization 7 2.2.3. X-ray production volume 10 2.2.4. Bremsstrahlung or continuum x-ray generation 11 2.2.5. Cathodoluminescence 12 3. QUANTITATIVE X-RAY SPECTROMETRY 13 3.1. MATRIX CORRECTIONS 14 3.1.1. Atomic number correction (Z) 14 3.1.2. Absorption correction (A) 16 3.1.3. -

Electron Probe Microanalysis (EPMA) in the Earth Sciences
This is a repository copy of Electron probe microanalysis (EPMA) in the Earth Sciences. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/135874/ Version: Accepted Version Proceedings Paper: Walshaw, R orcid.org/0000-0002-8319-9312 (Accepted: 2018) Electron probe microanalysis (EPMA) in the Earth Sciences. In: Book of Tutorials and Abstracts. Microbeam Analysis in the Earth Sciences, 13th EMAS Regional Workshop, 04-07 Sep 2018, University of Bristol. (In Press) Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ ELECTRON PROBE MICROANALYSIS (EPMA) IN THE EARTH SCIENCES R.D. Walshaw University of Leeds, School of Earth & Environment, Leeds Electron Microscopy & Spectroscopy Centre Woodhouse Lane, Leeds LS2 9JT, Great Britain e-mail: [email protected] 75 1. INTRODUCTION Electron probe microanalysis (EPMA) is a non-destructive, standards-based microanalytical technique, yielding fully quantitative chemical analyses of solid materials at the micron scale. -

Recent Progress in Scanning Electron Microscopy for the Characterization of fine Structural Details of Nano Materials
Progress in Solid State Chemistry 42 (2014) 1e21 Contents lists available at ScienceDirect Progress in Solid State Chemistry journal homepage: www.elsevier.com/locate/pssc Recent progress in scanning electron microscopy for the characterization of fine structural details of nano materials Mitsuo Suga a,*, Shunsuke Asahina a, Yusuke Sakuda a, Hiroyoshi Kazumori a, Hidetoshi Nishiyama a, Takeshi Nokuo a, Viveka Alfredsson b, Tomas Kjellman b, Sam M. Stevens c, Hae Sung Cho d, Minhyung Cho d, Lu Han e, Shunai Che e, Michael W. Anderson f, Ferdi Schüth g, Hexiang Deng h, Omar M. Yaghi i, Zheng Liu j, Hu Young Jeong k, Andreas Stein l, Kazuyuki Sakamoto m, Ryong Ryoo n,o, Osamu Terasaki d,p,** a JEOL Ltd., SM Business Unit, Tokyo, Japan b Physical Chemistry, Lund University, Sweden c Private Contributor, UK d Graduate School of EEWS, KAIST, Republic of Korea e School of Chemistry & Chemical Engineering, Shanghai Jiao Tong University, China f Centre for Nanoporous Materials, School of Chemistry, University of Manchester, UK g Department of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Mülheim, Germany h College of Chemistry and Molecular Sciences, Wuhan University, China i Department of Chemistry, University of California, Berkeley, USA j Nanotube Research Center, AIST, Tsukuba, Japan k UNIST Central Research Facilities/School of Mechanical & Advanced Materials Engineering, UNIST, Republic of Korea l Department of Chemistry, University of Minnesota, Minneapolis, USA m Department of Nanomaterials Science, Chiba University, -

Transmission Electron Microscope
GG 711: Advanced Techniques in Geophysics and Materials Science Lecture 14 Transmission Electron Microscope Pavel Zinin HIGP, University of Hawaii, Honolulu, USA www.soest.hawaii.edu\~zinin Resolution of SEM Transmission electron microscopy (TEM) is a microscopy technique whereby a beam of electrons is transmitted through an ultra thin specimen, interacting with the specimen as it passes through. An image is formed from the interaction of the electrons transmitted through the specimen; the image is magnified and focused onto an imaging device, such as a fluorescent screen, on a layer of photographic film, or to be detected by a sensor such as a CCD camera. The first TEM was built by Max Kroll and Ernst Ruska in 1931, with this group developing the first TEM with resolution power greater than that of light in 1933 and the first commercial TEM in 1939. The first practical TEM, Originally installed at I. G Farben-Werke and now on display at the Deutsches Museum in Munich, Germany Comparison of LM and TEM Both glass and EM lenses subject to same distortions and aberrations Glass lenses have fixed focal length, it requires to change objective lens to change magnification. We move objective lens closer to or farther away from specimen to focus. EM lenses to specimen distance fixed, focal length varied by varying current through lens LM: (a) Direct observation TEM: (a) Video imaging (CRT); (b) image of the image; (b) image is is formed by transmitted electrons impinging formed by transmitted light on phosphor coated screen Transmission Electron Microscope: Principle Ray diagram of a conventional transmission electron microscope (top Positioning of signal path) and of a scanning transmission electron microscope (bottom path). -

Light and Electron Microscopy of the Exocrine Pancreas in the Chronically Reserpinized Rat1
003 1 -3998/89/2505-0482$02.00/0 PEDIATRIC RESEARCH Vol. 25, No. 5, 1989 Copyright O 1989 International Pediatric Research Foundation, Inc. Printed rn U.S.A. Light and Electron Microscopy of the Exocrine Pancreas in the Chronically Reserpinized Rat1 GILLES GRONDIN, FRANCOIS A. LEBLOND, JEAN MORISSET, AND DENIS LEBEL Centre de Recherche sur Ies Micanismes de Sicrition, Faculty of Science, University of Sherbrooke, Sherbrooke, QC, Canada, JIK 2RI ABSTRACT. The effects of reserpine injections were stud- been the most studied (1-8). The main effects induced by the ied on the morphology of the pancreas in an experimental reserpine treatment on the ultrastructure of the pancreas are the model for cystic fibrosis, the chronically reserpinized rat. following: an accumulation of granules in the acinar cell, an A detailed examination of the tissue was carried out at the alteration of the granule ultrastructure, a slight diminution of light and electron microscopic levels. The nonspecific ef- the RER and Golgi apparatus, the presence of autophagic bodies, fects of secondary malnutrition induced by the drug were and an augmentation of lysosomal bodies (3,5,9, 10). In another assessed with a group of animals pair fed with the treated paper (1 I), we report the effects of such a treatment on pancreatic animals. In a companion paper, we show that pancreatic growth and on the amount of a glycoprotein characteristic of the wt, lipase, and GP-2 contents also are affected by reserpine zymogen granule membrane, the GP-2. In this study, we specif- treatment. In this study, we report that no morphologic ically report the effects of chronic reserpine treatment at two differences were observed between the exocrine pancreatic doses, on the structure and ultrastructure of the rat pancreas. -

The Future of Electron Microscopy
BNL-108108-2015-JA Feature Article for Physics Today The Future of Electron Microscopy Yimei Zhu and Hermann Dürr Seeing is believing. So goes the old adage, and seen evidence is undoubtedly satisfying in that it can be interpreted readily, though not necessarily always correctly. For centuries, humans have tried continuously to improve their abilities of seeing things, from developing telescopes to observe our solar systems, to microscopes to reveal bacteria and viruses. The 2014 Nobel Prize in Chemistry was awarded to Eric Betzig, Stefan Hell and William Moerner for their pioneering work on super-resolution fluorescence microscopy, overcoming Abbe’s diffraction limit in the resolution of conventional light microscopes. It was a breakthrough that enabled ground-breaking discoveries in biological research and also is testimony of the importance of modern microscopy. In February 2014 the US Department of Energy’s Office of Basic Energy Science held a two day workshop that brought together experts from various fields to identify new science that will be enabled by advances in electron microscopy. [1] The workshop focused on studies of ultrafast processes, atomic resolution, sample environments and measuring functionality at nanoscales. This article captures some of the exciting discussions the workshop generated. Seeing with electrons Electrons, photons and neutrons are three fundamental probes of condensed matter. They are routinely used to study complementary physical properties of materials. Neutrons interact with nuclei and atomic spins, x-ray photons with electron clouds and electron probes with electrostatic potentials, i.e., positively charged nuclei screened by negatively charged electrons. Compared with x-rays and neutrons, one advantage of electrons is their much stronger interaction with matter (105 times stronger than elastically scattered x-rays due to the coulomb interaction); hence, for a very small probing volume, the interaction generates strong signals, making it far easier to image and detect individual atoms, molecules, and nanoscale objects. -

Super-Resolution Imaging by Dielectric Superlenses: Tio2 Metamaterial Superlens Versus Batio3 Superlens
hv photonics Article Super-Resolution Imaging by Dielectric Superlenses: TiO2 Metamaterial Superlens versus BaTiO3 Superlens Rakesh Dhama, Bing Yan, Cristiano Palego and Zengbo Wang * School of Computer Science and Electronic Engineering, Bangor University, Bangor LL57 1UT, UK; [email protected] (R.D.); [email protected] (B.Y.); [email protected] (C.P.) * Correspondence: [email protected] Abstract: All-dielectric superlens made from micro and nano particles has emerged as a simple yet effective solution to label-free, super-resolution imaging. High-index BaTiO3 Glass (BTG) mi- crospheres are among the most widely used dielectric superlenses today but could potentially be replaced by a new class of TiO2 metamaterial (meta-TiO2) superlens made of TiO2 nanoparticles. In this work, we designed and fabricated TiO2 metamaterial superlens in full-sphere shape for the first time, which resembles BTG microsphere in terms of the physical shape, size, and effective refractive index. Super-resolution imaging performances were compared using the same sample, lighting, and imaging settings. The results show that TiO2 meta-superlens performs consistently better over BTG superlens in terms of imaging contrast, clarity, field of view, and resolution, which was further supported by theoretical simulation. This opens new possibilities in developing more powerful, robust, and reliable super-resolution lens and imaging systems. Keywords: super-resolution imaging; dielectric superlens; label-free imaging; titanium dioxide Citation: Dhama, R.; Yan, B.; Palego, 1. Introduction C.; Wang, Z. Super-Resolution The optical microscope is the most common imaging tool known for its simple de- Imaging by Dielectric Superlenses: sign, low cost, and great flexibility. -

The Chemical Activators of Cathodoluminescence in Jadeite
The Chemical Activators of Cathodoluminescence in Jadeite Erin C. Dopfel A thesis presented to the faculty of Mount Holyoke College in partial fulfillment of the requirements for the degree of Bachelor of Arts with Honor Department of Earth and Environment Mount Holyoke College South Hadley, Massachusetts May 2006 Thesis Advisor: Dr. M. Darby Dyar Department Chair: Dr. Mark McMenamin ACKNOWLEDGMENTS I would like to thank my advisor, Darby Dyar, for her patience and guidance, and all the support she has shown toward this project over the past year. I am also grateful to Dean Connie Allen and Mark McMenamin, members of my thesis committee, for reading this document and providing me with extraordinarily helpful suggestions. And to the faculty of Mount Holyoke’s Department of Earth and Environment, especially Michelle Markley, Al Werner, and Steve Dunn, who have acted not only as mentors, but as true friends. Endless thanks to the donors of the Mount Holyoke Summer Research Fellowship, the Martha Godchaux Field Scholarship, and the Department of Earth and Environment funds, as well as Darby Dyar and the Sorena Sorensen, for supporting my travels and research expenses. Thanks to Mike Jercinovic at the University of Massachusetts – Amherst Microprobe Lab, who somehow managed to run all of my EPMA analyses on the shortest-notice-known-to-man. I am especially grateful to William F. McDonough and Richard Ash at the University of Maryland’s Isotope Geochemistry Lab, for allowing me to utilize their facilities to collect my LA ICP-MS data. Also, thanks to Gerard Marchand, Yarrow Rothstein, and Eli Sklute of Mount Holyoke College for helping to prepare and change ii samples, and fit messy Mössbauer curves. -

Development of Optical Hyperlens for Imaging Below the Diffraction Limit
Development of optical hyperlens for imaging below the diffraction limit Hyesog Lee, Zhaowei Liu, Yi Xiong, Cheng Sun and Xiang Zhang* 5130 Etcheverry Hall, NSF Nanoscale Science and Engineering Center (NSEC), University of California, Berkeley, CA 94720 *Corresponding author: [email protected] http://xlab.me.berkeley.edu Abstract: We report here the design, fabrication and characterization of optical hyperlens that can image sub-diffraction-limited objects in the far field. The hyperlens is based on an artificial anisotropic metamaterial with carefully designed hyperbolic dispersion. We successfully designed and fabricated such a metamaterial hyperlens composed of curved silver/alumina multilayers. Experimental results demonstrate far-field imaging with resolution down to 125nm at 365nm working wavelength which is below the diffraction limit. ©2007 Optical Society of America OCIS codes: (110 0180) Microscopy; (220.4241) Optical design and fabrication: Nanostructure fabrication References and links 1. E. Abbe, Arch. Mikroskop. Anat. 9, 413 (1873) 2. E. Betzig, J. K. Trautman, T. D. Harris, J. S. Weiner and R. L. Kostelak, “Breaking the diffraction barrier – optical microscopy on a nanometric scale,” Science 251, 1468-1470 (1991) 3. S. W. Hell, “Toward Fluorescence nanoscopy,” Nat. Biotechnol. 21, 1347-1355 (2003) 4. M. G. L. Gustafsson, “Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution,” P. Natl. Acad. Sci. 102, 13081-13086 (2005) 5. J. B. Pendry, “Negative refraction makes a perfect lens,” Phys. Rev. Lett. 85, 3966-3969 (2000) 6. N. Fang, H. Lee, C. Sun and X. Zhang, “Sub-Diffraction-Limited Optical Imaging with a Silver Superlens” Science 308, 534-537 (2005) 7. -

Crystallography Beyond Crystals: PX and Spcryoem
GENERAL ARTICLE Crystallography beyond Crystals: PX and SPCryoEM Ramanathan Natesh Protein Crystallography (PX) or Macromolecular Crystal- lography has been unarguably one of the great intellectual achievements of 20th century. In fact, it has developed to be a strong base for ‘hybrid methods’ for macromolecular struc- ture determination by a combination of PX and Single Par- ticle Cryo Electron Microscopy (SPCryoEM). This article describes how PX and SPCryoEM are independent methods, how they contribute as hybrid methods, their complementarity Ramanathan Natesh is a Ramalingaswami Fellow – and supplementarity and their current status. This article DBT and Assistant Professor though not a complete review of PX or SPCryoEM of macro- at the School of Biology, molecular protein complexes, serves as a guideline in under- Indian Institute of Science standing the underlying principles. Education and Research, Thiruvananthapuram. His research interests involve 1. Introduction Molecular Structural Biology studies of proteins Despite the knowledge and popular use of PX techniques in the and its complexes in human scientific community, it is less known to the general public. The health and diseases using International Year of Crystallography (IYCr) 2014 is being orga- two principle techniques viz., Protein Crystallogra- nized jointly by the International Union of Crystallography (IUCr) phy and Single Particle and UNESCO, which has taken many measures to popularize Cryo Electron Microscopy crystallography. A UNESCO brochure on Crystallography Mat- along with range of other ters can be found here http://www.iycr2014.org/about/promo- biophysical and biochemical techniques. tional-materials. One question you might ask is, ‘why is knowing the protein 1 Inhibitors are also called as structure important?’ All living beings are made of cells and there drug molecules that stop the are thousands of proteins that function within and outside these function of the protein by bind- cells. -
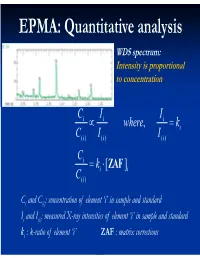
Electron Microprobe Analysis Slide 4
EPMA: Quantitative analysis WDS spectrum: Intensity is proportional to concentration C i I i Ii where, ki C( i )I i ( ) I() i C i ki [ ZAF i ] C( i ) Ci and C(i): concentration of element ‘i’ in sample and standard Ii and I(i): measured X-ray intensities of element ‘i’ in sample and standard ki : k-ratio of element ‘i’ ZAF : matrix corrections Matrix (ZAF) corrections Z : atomic number correction A : absorption correction F : fluorescence correction Atomic number (Z) correction R = CjRj Ri [ R = #X-ray photons S i generated / #photons if Z i * there were no back-scatter] Ri * S = C S S i j j [ S = -(1/)(dE/ds), * sample stopping power] Z, a function of E0 and composition Duncumb-Reed-Yakowitz method: Ri = j CjRij Rij = R' 1 - R'2 ln(R'3 Zj+25) -3 3 2 R'1 = 8.73x10 U - 0.1669 U + 0.9662 U + 0.4523 -3 3 -2 2 R'2 = 2.703x10 U - 5.182x10 U + 0.302 U - 0.1836 3 2 3 R'3 = (0.887 U - 3.44 U + 9.33 U - 6.43)/U Si = j CjSij Sij = (const) [(2Zj/Aj )/(E0+Ec)]ln[583(E0+Ec)/Jj ] where, J (keV) = (9.76Z + 58.82Z-0.19)x10-3 Z, a function of E0 and composition Al-Cu alloy 1.2 1.2 Pure metal standards Pure metal standards 1.15 1.15 1.1 1.1 1.05 1.05 1 1 AlK C CuK C 0.95 Cu 0.95 Cu Z 0.1 0.1 Z 0.9 0.3 0.9 0.3 0.5 0.5 0.85 0.7 0.85 0.7 0.9 0.9 0.8 0.8 10 15 20 10 15 20 E0 (keV) E0 (keV) 1.2 1.2 CuAl standard CuAl standard 1.15 2 1.15 2 1.1 1.1 1.05 1.05 1 1 AlK C CuK C 0.95 Cu 0.95 Cu Z 0.1 0.1 Z 0.9 0.3 0.9 0.3 0.5 0.5 0.85 0.7 0.85 0.7 0.9 0.9 0.8 0.8 10 15 20 10 15 20 E0 (keV) E0 (keV) X-ray absorption -(/)(x) -(/)( z cosec) I = I0 exp = I0 -

ANL-7917 Chemical Separations Processes for Plutonium and Uranium
ANL-7917 Chemical Separations Processes for Plutonium and Uranium ARGONNE NATIONAL LABORATORY 9700 South Cass Avenue Argonne, Illinois 60439 DEVELOPMENT STUDIES ON A FLUIDIZED-BED PROCESS FOR CONVERSION OF U/Pu NITRATES TO OXIDES Part 1. Laboratory-scale Denitration Studies by S. Vogler, D. E. Grosvenor, N. M. Levitz, and F. G. Teats Chemical Engineering Division April 1972 NOTICE- This report was prepared as an account of work sponsored by the United States Government. Neither the United States nor the United States Atomic Energy Commission, nor any of their employees, nor any of their contractors, subcontractors, or their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, com• pleteness or usefulness of any information, apparatus, product or process disclosed, or represents that its use would not infringe privately owned rights. BOTBimON DF THIS DOCUMENT IS ONLI DISCLAIMER This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency Thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.