ANL-7917 Chemical Separations Processes for Plutonium and Uranium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Electron Microprobe Analysis Course 12.141 Notes MIT Electron
Electron Microprobe Analysis Course 12.141 Notes Dr. Nilanjan Chatterjee The electron microprobe provides a complete micron-scale quantitative chemical analysis of inorganic solids. The method is nondestructive and utilizes characteristic x-rays excited by an electron beam incident on a flat surface of the sample. This course provides an introduction to the theory of x-ray microanalysis by wavelength and energy dispersive spectrometry (WDS and EDS), ZAF matrix correction procedures, and scanning electron imaging techniques using backscattered electron (BE), secondary electron (SE), x-ray (elemental mapping), and cathodoluminescence (CL) signals. Lab sessions involve hands- on use of the JEOL JXA-8200 Superprobe. MIT Electron Microprobe Facility Massachusetts Institute of Technology Department of Earth, Atmospheric & Planetary Sciences Room: 54-1221, Cambridge, MA 02139 Phone: (617) 253-1995; Fax: (617) 253-7102 e-mail: [email protected] web: http://web.mit.edu/e-probe/www/ Nilanjan ChatterjeeMIT, Cambridge, MA, USA October 2017 2 TABLE OF CONTENTS Page number 1. INTRODUCTION 3 2. ELECTRON SPECIMEN INTERACTIONS 5 2.1. ELASTIC SCATTERING 5 2.1.1. Electron backscattering 5 2.1.2. Electron interaction volume 6 2.2. INELASTIC SCATTERING 7 2.2.1. Secondary electron generation 7 2.2.2. Characteristic x-ray generation: inner-shell ionization 7 2.2.3. X-ray production volume 10 2.2.4. Bremsstrahlung or continuum x-ray generation 11 2.2.5. Cathodoluminescence 12 3. QUANTITATIVE X-RAY SPECTROMETRY 13 3.1. MATRIX CORRECTIONS 14 3.1.1. Atomic number correction (Z) 14 3.1.2. Absorption correction (A) 16 3.1.3. -

Electron Probe Microanalysis (EPMA) in the Earth Sciences
This is a repository copy of Electron probe microanalysis (EPMA) in the Earth Sciences. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/135874/ Version: Accepted Version Proceedings Paper: Walshaw, R orcid.org/0000-0002-8319-9312 (Accepted: 2018) Electron probe microanalysis (EPMA) in the Earth Sciences. In: Book of Tutorials and Abstracts. Microbeam Analysis in the Earth Sciences, 13th EMAS Regional Workshop, 04-07 Sep 2018, University of Bristol. (In Press) Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ ELECTRON PROBE MICROANALYSIS (EPMA) IN THE EARTH SCIENCES R.D. Walshaw University of Leeds, School of Earth & Environment, Leeds Electron Microscopy & Spectroscopy Centre Woodhouse Lane, Leeds LS2 9JT, Great Britain e-mail: [email protected] 75 1. INTRODUCTION Electron probe microanalysis (EPMA) is a non-destructive, standards-based microanalytical technique, yielding fully quantitative chemical analyses of solid materials at the micron scale. -

The Chemical Activators of Cathodoluminescence in Jadeite
The Chemical Activators of Cathodoluminescence in Jadeite Erin C. Dopfel A thesis presented to the faculty of Mount Holyoke College in partial fulfillment of the requirements for the degree of Bachelor of Arts with Honor Department of Earth and Environment Mount Holyoke College South Hadley, Massachusetts May 2006 Thesis Advisor: Dr. M. Darby Dyar Department Chair: Dr. Mark McMenamin ACKNOWLEDGMENTS I would like to thank my advisor, Darby Dyar, for her patience and guidance, and all the support she has shown toward this project over the past year. I am also grateful to Dean Connie Allen and Mark McMenamin, members of my thesis committee, for reading this document and providing me with extraordinarily helpful suggestions. And to the faculty of Mount Holyoke’s Department of Earth and Environment, especially Michelle Markley, Al Werner, and Steve Dunn, who have acted not only as mentors, but as true friends. Endless thanks to the donors of the Mount Holyoke Summer Research Fellowship, the Martha Godchaux Field Scholarship, and the Department of Earth and Environment funds, as well as Darby Dyar and the Sorena Sorensen, for supporting my travels and research expenses. Thanks to Mike Jercinovic at the University of Massachusetts – Amherst Microprobe Lab, who somehow managed to run all of my EPMA analyses on the shortest-notice-known-to-man. I am especially grateful to William F. McDonough and Richard Ash at the University of Maryland’s Isotope Geochemistry Lab, for allowing me to utilize their facilities to collect my LA ICP-MS data. Also, thanks to Gerard Marchand, Yarrow Rothstein, and Eli Sklute of Mount Holyoke College for helping to prepare and change ii samples, and fit messy Mössbauer curves. -
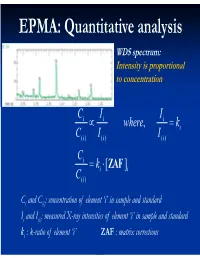
Electron Microprobe Analysis Slide 4
EPMA: Quantitative analysis WDS spectrum: Intensity is proportional to concentration C i I i Ii where, ki C( i )I i ( ) I() i C i ki [ ZAF i ] C( i ) Ci and C(i): concentration of element ‘i’ in sample and standard Ii and I(i): measured X-ray intensities of element ‘i’ in sample and standard ki : k-ratio of element ‘i’ ZAF : matrix corrections Matrix (ZAF) corrections Z : atomic number correction A : absorption correction F : fluorescence correction Atomic number (Z) correction R = CjRj Ri [ R = #X-ray photons S i generated / #photons if Z i * there were no back-scatter] Ri * S = C S S i j j [ S = -(1/)(dE/ds), * sample stopping power] Z, a function of E0 and composition Duncumb-Reed-Yakowitz method: Ri = j CjRij Rij = R' 1 - R'2 ln(R'3 Zj+25) -3 3 2 R'1 = 8.73x10 U - 0.1669 U + 0.9662 U + 0.4523 -3 3 -2 2 R'2 = 2.703x10 U - 5.182x10 U + 0.302 U - 0.1836 3 2 3 R'3 = (0.887 U - 3.44 U + 9.33 U - 6.43)/U Si = j CjSij Sij = (const) [(2Zj/Aj )/(E0+Ec)]ln[583(E0+Ec)/Jj ] where, J (keV) = (9.76Z + 58.82Z-0.19)x10-3 Z, a function of E0 and composition Al-Cu alloy 1.2 1.2 Pure metal standards Pure metal standards 1.15 1.15 1.1 1.1 1.05 1.05 1 1 AlK C CuK C 0.95 Cu 0.95 Cu Z 0.1 0.1 Z 0.9 0.3 0.9 0.3 0.5 0.5 0.85 0.7 0.85 0.7 0.9 0.9 0.8 0.8 10 15 20 10 15 20 E0 (keV) E0 (keV) 1.2 1.2 CuAl standard CuAl standard 1.15 2 1.15 2 1.1 1.1 1.05 1.05 1 1 AlK C CuK C 0.95 Cu 0.95 Cu Z 0.1 0.1 Z 0.9 0.3 0.9 0.3 0.5 0.5 0.85 0.7 0.85 0.7 0.9 0.9 0.8 0.8 10 15 20 10 15 20 E0 (keV) E0 (keV) X-ray absorption -(/)(x) -(/)( z cosec) I = I0 exp = I0 -

Electron Probe Microanalysis
Electron probe microanalysis Essential Knowledge Briefings First Edition, 2015 2 ELECTRON PROBE MICROANALYSIS Front cover image: high-resolution X-ray maps of copper (Cu), zinc (Zn) and silver (Ag) illustrating the interdiffusion zone between the main material (Cu) and the brazing material (Ag-Zn). Data acquired with the SXFiveFE at 10keV, 30nA © 2015 John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, UK Microscopy EKB Series Editor: Dr Julian Heath Spectroscopy and Separations EKB Series Editor: Nick Taylor ELECTRON PROBE MICROANALYSIS 3 CONTENTS 4 INTRODUCTION 6 HISTORY AND BACKGROUND 13 IN PRACTICE 22 PROBLEMS AND SOLUTIONS 28 WHAT’S NEXT? About Essential Knowledge Briefings Essential Knowledge Briefings, published by John Wiley & Sons, comprise a series of short guides to the latest techniques, appli - cations and equipment used in analytical science. Revised and updated annually, EKBs are an essential resource for scientists working in both academia and industry looking to update their understanding of key developments within each specialty. Free to download in a range of electronic formats, the EKB range is available at www.essentialknowledgebriefings.com 4 ELECTRON PROBE MICROANALYSIS INTRODUCTION Electron probe microanalysis (EPMA) is an analytical technique that has stood the test of time. Not only is EPMA able to trace its origins back to the discovery of X-rays at the end of the nineteenth century, but the first commercial instrument appeared over 50 years ago. Nevertheless, EPMA remains a widely used technique for determining the elemental composition of solid specimens, able to produce maps showing the distribution of elements over the surface of a specimen while also accurately measuring their concentrations. -

The Electron Microprobe Laboratory at Arizona State University
47th Lunar and Planetary Science Conference (2016) 3018.pdf THE ELECTRON MICROPROBE LABORATORY AT ARIZONA STATE UNIVERSITY. A. Wittmann1, D. Convey1, T. Sharp1, M. Wadhwa2, P. Buseck3, and K. Hodges4 1LeRoy Eyring Center for Solid State Science, Arizona State University, Box 871704, Tempe, AZ 85287-1704, [email protected]; 2Center for Meteorite Studies-School of Earth and Space Exploration, Arizona State University, Box 871404, Tempe, AZ 85287; 3School of Molecular Sciences, Arizona State University, Box 871604, Tempe, AZ 85287-1604; 4School of Earth and Space Exploration, Arizona State University, Box 876004, Tempe, AZ 85287-6004. Introduction: In 2011, Arizona State University For instrument control, we can utilize the custom- replaced their 25 year old electron microprobe with a ized JEOL system software and the PC-based Probe state-of-the-art JEOL JXA-8530F field emission in- for Windows software. strument. Our facility was partially funded by NASA Summary Analytical Capabilities: The electron and serves as an investigator facility instrument for microprobe laboratory at Arizona State University NASA’s Planetary Science Research Program. allows to determine the chemical composition of solid Technical Capacities: The JEOL JXA-8530F materials up to 10 cm in size that are stable under high HyperProbe is an electron microscope that has a nomi- vacuum. The non-destructive analytical technique does nal imaging resolution of 3 nm with a X-ray spectrom- not require special sample preparation and produces eter for non-destructive X-ray -

Scanning Electron Probe Microanalysis
1 Reference book not to be 1 1967 NBS TECHNICAL NOTE 278 Scanning Electron Probe Microanalysis KURT F. J. HEINRICH 7 > Si,? o U.S. DEPARTMENT OF COMMERCE 10 National Bureau of Standards Q \ ^ **CAU 0? — THE NATIONAL BUREAU OF STANDARDS The National Bureau of Standards 1 provides measurement and technical information services essential to the efficiency and effectiveness of the work of the Nation's scientists and engineers. The Bureau serves also as a focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. To accomplish this mission, the Bureau is organized into three institutes covering broad program areas of research and services: THE INSTITUTE FOR BASIC STANDARDS . provides the central basis within the United States for a complete and consistent system of physical measurements, coordinates that system with the measurement systems of other nations, and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. This Institute comprises a series of divisions, each serving a classical subject matter area: —Applied Mathematics—Electricity—Metrology—Mechanics—Heat—Atomic Physics—Physical Chemistry—Radiation Physics—Laboratory Astrophysics 2—Radio Standards Laboratory, 2 which includes Radio Standards Physics and Radio Standards Engineering—Office of Standard Refer- ence Data. THE INSTITUTE FOR MATERIALS RESEARCH . conducts materials research and provides associated materials services including mainly reference materials and data on the properties of ma- terials. Beyond its direct interest to the Nation's scientists and engineers, this Institute yields services which are essential to the advancement of technology in industry and commerce. -

A Simple Correction Procedure for Quantitative Electron Probe Microanalysis
' 'EFERENi UNITED STATES PARTMENT OF 3MMERCE NBS TECHNICAL NOTE 719 JBLICATION ^O'Co. '^rts o« A Simple Correction Procedure For Quantitative Electron Probe Microanalysis U.S. >ARTMENT OF OMMERCE 100 U5753 NATIONAL BUREAU OF STANDARDS 1 The National Bureau of Standards was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measure- ment system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Center for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scien- tific community, industry, and commerce. The Institute consists of a Center for Radia- tion Research, an Office of Measurement Services and the following divisions: Applied Mathematics—Electricity—Heat—Mechanics—Optical Physics—Linac Radiation -

Tutorial Review: X-Ray Mapping in Electron-Beam Instruments
Microsc. Microanal. 12, 2–25, 2006 / DOI: 10.1017 S1431927606060211 Microscopy AND Microanalysis © MICROSCOPY SOCIETY OF AMERICA 2006 Tutorial Review X-ray Mapping in Electron-Beam Instruments John J. Friel1 and Charles E. Lyman2,* 1Princeton Gamma Tech, C/N 863, Princeton, NJ 08542, USA 2Department of Materials Science and Engineering, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015, USA Abstract: This review traces the development of X-ray mapping from its beginning 50 years ago through current analysis procedures that can reveal otherwise obscure elemental distributions and associations. X-ray mapping or compositional imaging of elemental distributions is one of the major capabilities of electron beam microanalysis because it frees the operator from the necessity of making decisions about which image features contain elements of interest. Elements in unexpected locations, or in unexpected association with other elements, may be found easily without operator bias as to where to locate the electron probe for data collection. X-ray mapping in the SEM or EPMA may be applied to bulk specimens at a spatial resolution of about 1 mm. X-ray mapping of thin specimens in the TEM or STEM may be accomplished at a spatial resolution ranging from 2 to 100 nm, depending on specimen thickness and the microscope. Although mapping has traditionally been considered a qualitative technique, recent developments demonstrate the quantitative capabilities of X-ray mapping techniques. Moreover, the long-desired ability to collect and store an entire spectrum at every pixel is now a reality, and methods for mining these data are rapidly being developed. Key words: X-ray mapping, compositional imaging, X-ray spectrometry, EDS, WDS, concentration–concentration histograms, spectrum imaging, position-tagged spectrometry, principal component analysis, multivariate statis- tical analysis INTRODUCTION cused to a small probe, they can be deflected to form a scanned beam raster, and they can excite atoms in the specimen to produce characteristic X-ray signals. -

Comparison of Secondary Ion Mass Spectrometry (Sims) with Electron Microprobe Analysis (Epma) and Other Thin Film Analytical Methods H
COMPARISON OF SECONDARY ION MASS SPECTROMETRY (SIMS) WITH ELECTRON MICROPROBE ANALYSIS (EPMA) AND OTHER THIN FILM ANALYTICAL METHODS H. Werner, A. von Rosenstiel To cite this version: H. Werner, A. von Rosenstiel. COMPARISON OF SECONDARY ION MASS SPECTROME- TRY (SIMS) WITH ELECTRON MICROPROBE ANALYSIS (EPMA) AND OTHER THIN FILM ANALYTICAL METHODS. Journal de Physique Colloques, 1984, 45 (C2), pp.C2-103-C2-113. 10.1051/jphyscol:1984225. jpa-00223938 HAL Id: jpa-00223938 https://hal.archives-ouvertes.fr/jpa-00223938 Submitted on 1 Jan 1984 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. JOURNAL DE PHYSIQUE Colloque C2, supplément au n°2, Tome 45, février 198* page C2-103 COMPARISON OF SECONDARY ION MASS SPECTROMETRY (SIMS) WITH ELECTRON MICROPROBE ANALYSIS (EPMA) AND OTHER THIN FILM ANALYTICAL METHODS H.W. Werner and A.P. von Rosenstiel* Philips Research Laboratories, NL-5600 JA Eindhoven, The Netherlands Metaalinstituut TNO, NL-7300 AM Apeldoorn, The Netherlands Résumé - On décrit différents modes de SIMS pour l'analyse de couches minces. Le principe de la technique est suivi d'une discussion de certains aspects relatifs à l'instrumentation : le type de sources ioniques et leurs caracté ristiques, les avantages comparés de la microsonde ionique et du microscope ionique, les modes spéciaux de SIMS, la spectrométrie de masse a pulvérisa tion par particules neutres (SNMS) et le bombardement par atomes rapides (FAB). -

Energy-Dispersive X-Ray Microanalysis
ENERGY-DISPERSIVE X-RAY MICROANALYSIS An Introduction NORAN Instruments Middleton, Wisconsin Copyright 1983, 1988, 1989 Kevex Instruments, Inc., 1999 NORAN Instruments. All Rights Reserved. NORAN Instruments 2551 West Beltline Highway Middleton, WI 53562 +1 (608) 831-6511 http://www.noran.com Contents Preface vii Aside: A Little History 3 2.1 Electron-Sample Interactions 6 2.1.1 Secondary Electrons 6 2.1.2 Backscattered Electrons 7 2.1.3 X-Ray Continuum 7 2.1.4 Characteristic X-Rays 8 Nomenclature 9 Moseley’s Law 10 Characteristic X-Ray Intensity 10 2.2 Auger Electron Emission 11 2.3 Photon-Specimen Interactions 11 2.3.1 Absorption 11 2.3.2 Secondary Fluorescence 12 3.1 Beam Current 14 3.2 Accelerating Voltage 14 3.3 Beam-Specimen-Detector Geometry 15 3.3.1 Solid Angle 15 3.3.2 Take-off Angle 15 3.3.3 Incidence Angle 16 3.4 Vacuum Systems and Contamination 17 4.1 The Detector 19 4.1.1 The Physics of X-Ray Detection 20 4.1.2 Leakage Current and Lithium Drifting 20 4.1.3 Spectral Resolution 21 4.1.4 Detector Efficiency 21 4.1.5 The Dead Layer 22 4.1.6 Escape Peaks 22 4.2 Preamplifier 23 v Contents 4.3 Pulse Processor/Amplifier 24 4.3.1 Time-Variant Processing 24 4.3.2 Pulse Pileup Rejection 24 4.4 EDC and Multichannel Analyzer 25 Aside: Statistical Considerations 26 Normal Distribution and Standard Deviation 26 Counting Error 27 Minimum Detection Limits 29 5.1 Qualitative Analysis 34 5.1.1 Removing Escape Peaks 34 5.1.2 Peak Overlap 34 5.1.3 Effect Accelerating Voltage 35 5.1.4 Line Profiles, Dot Maps, and Spatial Resolution 35 5.2 Quantitative -

Characterization of Airborne a Nuclear Fuel
D P-1470 Distribution Category: UC-11 CHARACTERIZATION OF AIRBORNE PLUTONIUM-BEARING PARTICLES FROM A NUCLEAR FUEL REPROCESSING PLANT by S. Marshall Sanders, Jr. Approved by E. L. Albenesius, Research Manager Environmental Effects Division Pub1 ication Date: November 1977 NOTICE Thir mprt was prepared as an account of work sponsored by the United Stater Govemmnt. Neither the Umtcd Slates nor the United Stater Lkpartment of Energy, nor any of their employees. nor any of thcu contractors, rubcontractorr. or their employees, makes any wuarranty. cxprers or implied. or murnes any legal liabllity or rerponnbllity for the accuracy. complctcncrr 01 uvfulncrr of any miormt~on.apparatus. product or proas discloud, or reprrwnrr that its UIL would hot lnlrmge privately owmd nghu. I E. I. DU PONT DE NEMOURS AND COMPANY SAVANNAH RIVER LABORATORY AIKEN, SOUTH CAROLINA 29801 PREPARED FOR THE U.S. DEPARTMENT OF ENERGY UNDER CONTRACT AT(07-2)-1 This document is - Date:& /2047 DISCLAIMER This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency Thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof.