Molecular Diagnosis of Fanconi Anemia with Next-Generation Sequencing in a Case with Subtle Signs and a Negative Chromosomal Breakage Test
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -

Methemoglobinemia and Ascorbate Deficiency in Hemoglobin E Β Thalassemia: Metabolic and Clinical Implications
From www.bloodjournal.org by guest on April 2, 2015. For personal use only. Plenary paper Methemoglobinemia and ascorbate deficiency in hemoglobin E  thalassemia: metabolic and clinical implications Angela Allen,1,2 Christopher Fisher,1 Anuja Premawardhena,3 Dayananda Bandara,4 Ashok Perera,4 Stephen Allen,2 Timothy St Pierre,5 Nancy Olivieri,6 and David Weatherall1 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom; 2College of Medicine, Swansea University, Swansea, United Kingdom; 3University of Kelaniya, Colombo, Sri Lanka; 4National Thalassaemia Centre, District Hospital, Kurunegala, Sri Lanka; 5School of Physics, University of Western Australia, Crawley, Australia; and 6Hemoglobinopathy Research, University Health Network, Toronto, ON During investigations of the phenotypic man hypoxia induction factor pathway is There was, in addition, a highly signifi- diversity of hemoglobin (Hb) E  thalasse- not totally dependent on ascorbate lev- cant correlation between methemoglobin mia, a patient was encountered with per- els. A follow-up study of 45 patients with levels, splenectomy, and factors that sistently high levels of methemoglobin HbE  thalassemia showed that methemo- modify the degree of globin-chain imbal- associated with a left-shift in the oxygen globin levels were significantly increased ance. Because methemoglobin levels are dissociation curve, profound ascorbate and that there was also a significant re- modified by several mechanisms and may deficiency, and clinical features of scurvy; duction in plasma ascorbate levels. Hap- play a role in both adaptation to anemia these abnormalities were corrected by toglobin levels were significantly re- and vascular damage, there is a strong treatment with vitamin C. -
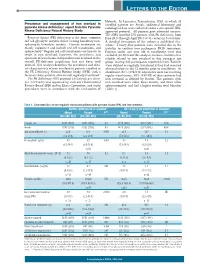
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin. -

The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2
CASE REPORT The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2 1Department of Medicine, Saint Mary Hospital, Nakhon Ratchasima 30000, Thailand, 2Department of General Practice, NopparatRajathanee Hospital, Khanna Yao, Bangkok 10230, Thailand ABSTRACT Polycythemia vera (PV) is a clonal myeloproliferative neoplasm mainly characterized by an abnormal increase of erythroid precursor cells leading to increased red blood cells (RBC) production that is opposite to iron deficiency anemia (IDA) of which the RBC production is decreased due to iron deficiency. This report was aimed to present one patient who had coexistence of these two opposite entities of the RBC production. She was a 47-year-old Thai who was admitted because of acute coronary syndrome and she was accidentally found to have microcytosis of RBC despite normal hemoglobin (Hb) concentration, Hb 14.7 g%, mean corpuscular volume (MCV) 70.0 fL, white blood cells 12,400/mm3, and platelet 401,000/mm3. The Hb analysis showed only A2A, with normal Hb A2 percentage. The polymerase chain reaction for alpha thalassemia-1 genotype was tested negative. Due to neither alpha- nor beta-thalassemia trait detected, the iron study was performed: Serum ferritin 6.1 ng/mL, serum iron 64 ug/dl, and total iron binding capacity 198 ug/dl. The iron storage was seemingly insufficient; hence, iron supplement was started and continued for 4 months. Her blood tests showed: Hb 18.3 g%, MCV 87.2 fl, serum ferritin 31.7 ng/ml, erythropoietin <1 IU/l, positive JAK2 V617F mutation, and normal oxygen saturation. The diagnosis of PV was definitely concluded and she was finally treated with hydroxyurea and occasional phlebotomy. -

Congenital Methemoglobinemia Identified by Pulse Oximetry Screening Jennifer Ward, Jayashree Motwani, Nikki Baker, Matthew Nash, Andrew K
Congenital Methemoglobinemia Identified by Pulse Oximetry Screening Jennifer Ward, BMBS,a Jayashree Motwani, MBBS,b Nikki Baker, MSc,a Matthew Nash, MBChB,a Andrew K. Ewer, MD,a,c Gergely Toldi, MDa Congenital methemoglobinemia is a rare condition caused by cytochrome b5 abstract reductase deficiency, cytochrome b5 deficiency, or hemoglobin M disease. Newborn pulse oximetry screening was developed for the early detection of critical congenital heart disease; however, it also enables the early identification of other hypoxemic conditions. We present the case of a term neonate who was admitted to the neonatal unit after a failed pulse oximetry screening at 3 hours of age. Oxygen saturations remained between 89% and 92% despite an increase in oxygen therapy. Chest radiograph and echocardiogram results were normal. A capillary blood gas test had normal results except for a raised methemoglobin level of 16%. Improvement was Departments of aNeonatology and bHaematology, seen on the administration of methylene blue, which also resulted in an Birmingham Women’s and Children’s Hospital, Birmingham, increase in oxygen saturations to within normal limits. Further investigation United Kingdom; and cInstitute of Metabolism and Systems Research, University of Birmingham, Birmingham, United revealed evidence of type I hereditary cytochrome b5 reductase deficiency as Kingdom a result of a CYB5R3 gene mutation with 2 pathogenic variants involving Drs Ward and Toldi were responsible for neonatal guanine-to-adenine substitutions. Although mild cyanosis is generally the care and drafted and reviewed the manuscript; Dr only symptom of type I disease, patients may later develop associated Nash and Ms Baker were responsible for neonatal symptoms, such as fatigue and shortness of breath. -

Hemoglobin Bart's and Alpha Thalassemia Fact Sheet
Health Care Provider Hemoglobinopathy Fact Sheet Hemoglobin Bart’s & Alpha Thalassemia Hemoglobin Bart’s is a tetramer of gamma (fetal) globin chains seen during the newborn period. Its presence indicates that one or more of the four genes that produce alpha globin chains are dysfunctional, causing alpha thalassemia. The more alpha genes affected, the more significant the thalassemia and clinical symptoms. Alpha thalassemia occurs in individuals of all ethnic backgrounds and is one of the most common genetic diseases worldwide. However, the clinically significant forms (Hemoglobin H disease, Hemoglobin H Constant Spring, and Alpha Thalassemia Major) occur predominantly among Southeast Asians. Summarized below are the manifestations associated with the different levels of Hemoglobin Bart’s detected on the newborn screen, and recommendations for follow-up. The number of dysfunctional genes is estimated by the percentage of Bart’s seen on the newborn screen. Silent Carrier- Low Bart’s If only one alpha gene is affected, the other three genes can compensate nearly completely and only a low level of Bart’s is detected, unless hemoglobin Constant Spring is identified (see below). Levels of Bart’s below a certain percentage are not generally reported by the State Newborn Screening Program as these individuals are likely to be clinically and hematologically normal. However, a small number of babies reported as having possible alpha thalassemia trait will be silent carriers. Alpha Thalassemia or Hemoglobin Constant Spring Trait- Moderate Bart’s Alpha thalassemia trait produces a moderate level of Bart’s and typically results from the dysfunction of two alpha genes-- either due to gene deletions or a specific change in the alpha gene that produces elongated alpha globin and has a thalassemia-like effect: hemoglobin Constant Spring. -

Ineffective Erythropoiesis in -Thalassaemia
International Journal of Molecular Sciences Review Ineffective Erythropoiesis in β-Thalassaemia: Key Steps and Therapeutic Options by Drugs Filomena Longo *,† , Andrea Piolatto † , Giovanni Battista Ferrero and Antonio Piga Department of Clinical and Biological Sciences, University of Torino, 10043 Torino, Italy; [email protected] (A.P.); [email protected] (G.B.F.); [email protected] (A.P.) * Correspondence: fi[email protected]; Tel.: +39-0119026032 † These authors contributed equally to this work. Abstract: β-thalassaemia is a rare genetic condition caused by mutations in the β-globin gene that result in severe iron-loading anaemia, maintained by a detrimental state of ineffective erythropoiesis (IE). The role of multiple mechanisms involved in the pathophysiology of the disease has been recently unravelled. The unbalanced production of α-globin is a major source of oxidative stress and membrane damage in red blood cells (RBC). In addition, IE is tightly linked to iron metabolism dysregulation, and the relevance of new players of this pathway, i.e., hepcidin, erythroferrone, matriptase-2, among others, has emerged. Advances have been made in understanding the balance between proliferation and maturation of erythroid precursors and the role of specific factors in this process, such as members of the TGF-β superfamily, and their downstream effectors, or the transcription factor GATA1. The increasing understanding of IE allowed for the development of a broad set of potential therapeutic options beyond the current standard of care. Many candidates of disease-modifying drugs are currently under clinical investigation, targeting the regulation of iron metabolism, the production of foetal haemoglobin, the maturation process, or the energetic balance Citation: Longo, F.; Piolatto, A.; and membrane stability of RBC. -

Dysregulated Iron Metabolism in Polycythemia Vera: Etiology and Consequences
Leukemia (2018) 32:2105–2116 https://doi.org/10.1038/s41375-018-0207-9 REVIEW ARTICLE Chronic myeloproliferative neoplasms Dysregulated iron metabolism in polycythemia vera: etiology and consequences 1 1 1 2 3 1 Yelena Z. Ginzburg ● Maria Feola ● Eran Zimran ● Judit Varkonyi ● Tomas Ganz ● Ronald Hoffman Received: 17 May 2018 / Revised: 7 June 2018 / Accepted: 18 June 2018 / Published online: 24 July 2018 © The Author(s) 2018. This article is published with open access Abstract Polycythemia vera (PV) is a chronic myeloproliferative neoplasm. Virtually all PV patients are iron deficient at presentation and/or during the course of their disease. The co-existence of iron deficiency and polycythemia presents a physiological disconnect. Hepcidin, the master regulator of iron metabolism, is regulated by circulating iron levels, erythroblast secretion of erythroferrone, and inflammation. Both decreased circulating iron and increased erythroferrone levels, which occur as a consequence of erythroid hyperplasia in PV, are anticipated to suppress hepcidin and enable recovery from iron deficiency. Inflammation which accompanies PV is likely to counteract hepcidin suppression, but the relatively low serum ferritin levels observed suggest that inflammation is not a major contributor to the dysregulated iron metabolism. Furthermore, potential fi 1234567890();,: 1234567890();,: defects in iron absorption, aberrant hypoxia sensing and signaling, and frequency of bleeding to account for iron de ciency in PV patients have not been fully elucidated. Insufficiently suppressed hepcidin given the degree of iron deficiency in PV patients strongly suggests that disordered iron metabolism is an important component of the pathobiology of PV. Normalization of hematocrit levels using therapeutic phlebotomy is the most common approach for reducing the incidence of thrombotic complications, a therapy which exacerbates iron deficiency, contributing to a variety of non-hematological symptoms. -

Patient History for Hemoglobinopathy
500 Chipeta Way Salt Lake City, UT 84108-1221 phone: 801-583-2787 | toll free: 800-242-2787 fax: 801-584-5249 | aruplab.com THIS IS NOT A TEST REQUEST FORM. Please complete and submit with the test request form or electronic packing list. PATIENT HISTORY FOR HEMOGLOBINOPATHY/THALASSEMIA TESTING Patient Name: Date of Birth: Sex: Female Male ☐ ☐ Ordering Provider: Provider’s Phone: Practice Specialty: Provider’s Fax: Genetic Counselor: Counselor Phone: Patient’s Ethnicity/Ancestry (check all that apply) African American/Black Asian Hispanic White Other: ☐ ☐ ☐ ☐ ☐ List country of origin (if known): Does the patient have symptoms? .................................................................. No Yes (check all that apply and describe) ☐ ☐ Anemia: Has iron deficiency been excluded? .......................................................................... No Yes Unknown ☐ ☐ ☐ ☐ Splenomegaly Other symptoms: ☐ ☐ Has the patient had a recent transfusion? ................... No Yes; date of transfusion: Unknown ☐ ☐ ☐ Laboratory Findings: (Indicate which testing was performed and provide results, as requested.) ☐ Hemoglobin evaluation by electrophoresis or HPLC; date performed: Hb A%: Hb C%: Hb F%: Other: Hb A2%: Hb E%: Hb S%: CBC: date performed: HGB: HCT: MCV: Reticulocyte count: ( %) ☐ Has the patient undergone previous DNA testing? ...................................................................... ☐ No Yes Unknown ☐ ☐ If yes, check the completed test(s) and provide the result or attach a copy of the laboratory report. Alpha globin deletion analysis; result: ☐ Beta globin sequencing; result: ☐ Other: ☐ Is there any relevant family history of hemoglobinopathy/thalassemia? ..................................... No Yes Unknown ☐ ☐ ☐ If yes, specify the relative's relationship to the patient: ; The relative is: a healthy carrier / affected ☐ ☐ List the gene and variant(s) identified or attach a copy of the relative's laboratory result: Check the test you intend to order. -

Alpha-Thalassemia
Alpha-thalassemia What is alpha-thalassemia? Alpha-thalassemia is an inherited disorder with variable severity. Individuals with alpha-thalassemia have a deficiency in the production of hemoglobin, which carries oxygen in the blood. Signs and symptoms of alpha-thalassemia are due to a reduction in the amount of hemoglobin that reaches the body’s tissues. There are two clinically significant forms of alpha thalassemia: HbH disease and Hb Bart hydrops fetalis syndrome.1 What are the symptoms of alpha-thalassemia and what treatment is available? Signs and symptoms of the more severe Hb Bart hydrops fetalis syndrome appear before birth and may include:1 Hydrops fetalis (abnormal fluid accumulation in the body before birth) Severe anemia Enlarged liver and spleen (hepatosplenomegaly) Heart problems Abnormalities of the urinary system or genitalia Signs and symptoms of the less severe HbH disease usually appear in early childhood and may include:1 Mild to moderate anemia with associated weakness and fatigue Enlarged liver and spleen (hepatosplenomegaly) Jaundice Bone changes In utero stem cell transplantation or fetal blood transfusions may be available for affected fetuses.3 Without treatment, most babies with Hb Bart hydrops fetalis syndrome are stillborn or die soon after birth. Mothers of babies affected with Hb Bart hydrops fetalis syndrome may experience serious pregnancy complications, including preeclampsia, premature delivery, or abnormal bleeding.1,3 Individuals with HbH disease have a shortened lifespan.1,2 Treatment is supportive and may include red blood cell transfusions.3 Alpha-thalassemia is included in newborn screening programs in all states in the United States.4 How is alpha-thalassemia inherited? Alpha-thalassemia is caused by mutations, usually deletions, in the alpha-globin gene cluster.1 Inheritance is usually autosomal recessive.3 Each person has two HBA genes, HBA1 and HBA2. -

Beta Thalassemia
Beta thalassemia Description Beta thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the iron-containing protein in red blood cells that carries oxygen to cells throughout the body. In people with beta thalassemia, low levels of hemoglobin lead to a lack of oxygen in many parts of the body. Affected individuals also have a shortage of red blood cells ( anemia), which can cause pale skin, weakness, fatigue, and more serious complications. People with beta thalassemia are at an increased risk of developing abnormal blood clots. Beta thalassemia is classified into two types depending on the severity of symptoms: thalassemia major (also known as Cooley's anemia) and thalassemia intermedia. Of the two types, thalassemia major is more severe. The signs and symptoms of thalassemia major appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged spleen, liver, and heart, and their bones may be misshapen. Some adolescents with thalassemia major experience delayed puberty. Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body, resulting in liver, heart, and hormone problems. Thalassemia intermedia is milder than thalassemia major. The signs and symptoms of thalassemia intermedia appear in early childhood or later in life.