Beta Brochure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phenotypic and Molecular Genetic Analysis of Pyruvate Kinase Deficiency in a Tunisian Family
The Egyptian Journal of Medical Human Genetics (2016) 17, 265–270 HOSTED BY Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com ORIGINAL ARTICLE Phenotypic and molecular genetic analysis of Pyruvate Kinase deficiency in a Tunisian family Jaouani Mouna a,1,*, Hamdi Nadia a,1, Chaouch Leila a, Kalai Miniar a, Mellouli Fethi b, Darragi Imen a, Boudriga Imen a, Chaouachi Dorra a, Bejaoui Mohamed b, Abbes Salem a a Laboratory of Molecular and Cellular Hematology, Pasteur Institute, Tunis, Tunisia b Service d’Immuno-He´matologie Pe´diatrique, Centre National de Greffe de Moelle Osseuse, Tunis, Tunisia Received 9 July 2015; accepted 6 September 2015 Available online 26 September 2015 KEYWORDS Abstract Pyruvate Kinase (PK) deficiency is the most frequent red cell enzymatic defect responsi- Pyruvate Kinase deficiency; ble for hereditary non-spherocytic hemolytic anemia. The disease has been studied in several ethnic Phenotypic and molecular groups. However, it is yet an unknown pathology in Tunisia. We report here, the phenotypic and investigation; molecular investigation of PK deficiency in a Tunisian family. Hemolytic anemia; This study was carried out on two Tunisian brothers and members of their family. Hematolog- Hydrops fetalis; ical, biochemical analysis and erythrocyte PK activity were performed. The molecular characteriza- PKLR mutation tion was carried out by gene sequencing technique. The first patient died few hours after birth by hydrops fetalis, the second one presented with neonatal jaundice and severe anemia necessitating urgent blood transfusion. This severe clinical pic- ture is the result of a homozygous mutation of PKLR gene at exon 8 (c.1079G>A; p.Cys360Tyr). -

Non-Commercial Use Only
only use Non-commercial 14th International Conference on Thalassaemia and Other Haemoglobinopathies 16th TIF Conference for Patients and Parents 17-19 November 2017 • Grand Hotel Palace, Thessaloniki, Greece only use For thalassemia patients with chronic transfusional iron overload... Make a lasting impression with EXJADENon-commercial film-coated tablets The efficacy of deferasirox in a convenient once-daily film-coated tablet Please see your local Novartis representative for Full Product Information Reference: EXJADE® film-coated tablets [EU Summary of Product Characteristics]. Novartis; August 2017. Important note: Before prescribing, consult full prescribing information. iron after having achieved a satisfactory body iron level and therefore retreatment cannot be recommended. ♦ Maximum daily dose is 14 mg/kg body weight. ♦ In pediatric patients the Presentation: Dispersible tablets containing 125 mg, 250 mg or 500 mg of deferasirox. dosing should not exceed 7 mg/kg; closer monitoring of LIC and serum ferritin is essential Film-coated tablets containing 90 mg, 180 mg or 360 mg of deferasirox. to avoid overchelation; in addition to monthly serum ferritin assessments, LIC should be Indications: For the treatment of chronic iron overload due to frequent blood transfusions monitored every 3 months when serum ferritin is ≤800 micrograms/l. (≥7 ml/kg/month of packed red blood cells) in patients with beta-thalassemia major aged Dosage: Special population ♦ In moderate hepatic impairment (Child-Pugh B) dose should 6 years and older. ♦ Also indicated for the treatment of chronic iron overload due to blood not exceed 50% of the normal dose. Should not be used in severe hepatic impairment transfusions when deferoxamine therapy is contraindicated or inadequate in the following (Child-Pugh C). -

Hb S/Beta-Thalassemia
rev bras hematol hemoter. 2 0 1 5;3 7(3):150–152 Revista Brasileira de Hematologia e Hemoterapia Brazilian Journal of Hematology and Hemotherapy www.rbhh.org Scientific Comment ଝ The compound state: Hb S/beta-thalassemia ∗ Maria Stella Figueiredo Universidade Federal de São Paulo (UNIFESP), São Paulo, SP, Brazil Sickle cell disease (SCD) results from a single amino acid small increase in Hb concentration and in packed cell volume. substitution in the gene encoding the -globin subunit However, these effects are not accompanied by any reduction  ( 6Glu > Val) that produces the abnormal hemoglobin (Hb) in vaso-occlusive events, probably due to the great number of named Hb S. SCD has different genotypes with substantial Hb S-containing red blood cells resulting in increased blood 1,2 4,5 variations in presentation and clinical course (Table 1). The viscosity. combination of the sickle cell mutation and beta-thalassemia A confusing diagnostic problem is the differentiation 0  (-Thal) mutation gives rise to a compound heterozygous con- of Hb S/ -Thal from sickle cell anemia associated with ␣ dition known as Hb S/ thalassemia (Hb S/-Thal), which was -thalassemia. Table 2 shows that hematologic and elec- 3 first described in 1944 by Silvestroni and Bianco. trophoretic studies are unable to distinguish between the two The polymerization of deoxygenated Hb S (sickling) is the conditions and so family studies and DNA analysis are needed 5 primary event in the molecular pathogenesis of SCD. How- to confirm the diagnosis. +  ever, this event is highly dependent on the intracellular Hb In Hb S/ -Thal, variable amounts of Hb A dilute Hb S and composition; in other words, it is dependent on the concen- consequently inhibit polymerization-induced cellular dam- tration of Hb S, and type and concentration of the other types age. -

Non-Transfusion-Dependent Thalassemias
REVIEW ARTICLES Non-transfusion-dependent thalassemias Khaled M. Musallam, 1 Stefano Rivella, 2 Elliott Vichinsky, 3 and Eliezer A. Rachmilewitz, 4 1Department of Medicine and Medical Specialties, IRCCS Ca’ Granda Foundation Maggiore Policlinico Hospital, University of Milan, Milan, Italy; 2Department of Pediatrics, Division of Hematology/Oncology, Weill Medical College of Cornell University, New York, NY, USA; 3Department of Hematology and Oncology, Children’s Hospital and Research Center Oakland, Oakland, CA, USA; and 4Department of Hematology, Wolfson Medical Center, Holon, Israel ABSTRACT Non-transfusion-dependent thalassemias include a variety of phenotypes that, unlike patients with beta ( β)-tha - lassemia major, do not require regular transfusion therapy for survival. The most commonly investigated forms are β-thalassemia intermedia, hemoglobin E/ β-thalassemia, and α-thalassemia intermedia (hemoglobin H disease). However, transfusion-independence in such patients is not without side effects. Ineffective erythropoiesis and peripheral hemolysis, the hallmarks of disease process, lead to a variety of subsequent pathophysiologies including iron overload and hypercoagulability that ultimately lead to a number of serious clinical morbidities. Thus, prompt and accurate diagnosis of non-transfusion-dependent thalassemia is essential to ensure early intervention. Although several management options are currently available, the need to develop more novel therapeutics is jus - tified by recent advances in our understanding of the mechanisms of disease. Such efforts require wide interna - tional collaboration, especially since non-transfusion-dependent thalassemias are no longer bound to low- and middle-income countries but have spread to large multiethnic cities in Europe and the Americas due to continued migration. Introduction survival, although they may require occasional or even fre - quent transfusions in certain clinical settings and usually for Inherited hemoglobin disorders can be divided into two defined periods of time (Figure 1). -

The Variable Manifestations of Disease in Pyruvate Kinase Deficiency and Their Management
REVIEW ARTICLE The variable manifestations of disease in pyruvate kinase deficiency and their Ferrata Storti Foundation management Hanny Al-Samkari,1 Eduard J. van Beers,2 Kevin H.M. Kuo,3 Wilma Barcellini,4 Paola Bianchi,4 Andreas Glenthøj,5 María del Mar Mañú Pereira,6 Richard van Wijk,7 Bertil Glader8 and Rachael F. Grace9 1Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; 2Van Creveldkliniek, University Medical Centre Utrecht, University of Utrecht, Utrecht, the Netherlands; 3Division of Hematology, University of Toronto, University Health Haematologica 2020 Network, Toronto, Ontario, Canada; 4UOS Ematologia, Fisiopatologia delle Anemie, Volume 105(9):2229-2239 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy; 5Department of Hematology, Herlev and Gentofte Hospital, Herlev, Denmark; 6Translational Research in Rare Anaemia Disorders, Vall d'Hebron Institut de Recerca (VHIR), Barcelona, Spain; 7Department of Clinical Chemistry & Hematology, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands; 8Lucile Packard Children’s Hospital, Stanford University School of Medicine, Palo Alto, CA, USA and 9Dana/Farber Boston Children's Cancer and Blood Disorders Center, Harvard Medical School, Boston, MA, USA. ABSTRACT yruvate kinase deficiency (PKD) is the most common cause of chron- ic hereditary non-spherocytic hemolytic anemia and results in a Pbroad spectrum of disease. The diagnosis of PKD requires a high index of suspicion and judicious use of laboratory tests that may not always be informative, including pyruvate kinase enzyme assay and genetic analysis of the PKLR gene. A significant minority of patients with PKD have occult mutations in non-coding regions of PKLR which are missed on standard genetic tests. -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -

Methemoglobinemia and Ascorbate Deficiency in Hemoglobin E Β Thalassemia: Metabolic and Clinical Implications
From www.bloodjournal.org by guest on April 2, 2015. For personal use only. Plenary paper Methemoglobinemia and ascorbate deficiency in hemoglobin E  thalassemia: metabolic and clinical implications Angela Allen,1,2 Christopher Fisher,1 Anuja Premawardhena,3 Dayananda Bandara,4 Ashok Perera,4 Stephen Allen,2 Timothy St Pierre,5 Nancy Olivieri,6 and David Weatherall1 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom; 2College of Medicine, Swansea University, Swansea, United Kingdom; 3University of Kelaniya, Colombo, Sri Lanka; 4National Thalassaemia Centre, District Hospital, Kurunegala, Sri Lanka; 5School of Physics, University of Western Australia, Crawley, Australia; and 6Hemoglobinopathy Research, University Health Network, Toronto, ON During investigations of the phenotypic man hypoxia induction factor pathway is There was, in addition, a highly signifi- diversity of hemoglobin (Hb) E  thalasse- not totally dependent on ascorbate lev- cant correlation between methemoglobin mia, a patient was encountered with per- els. A follow-up study of 45 patients with levels, splenectomy, and factors that sistently high levels of methemoglobin HbE  thalassemia showed that methemo- modify the degree of globin-chain imbal- associated with a left-shift in the oxygen globin levels were significantly increased ance. Because methemoglobin levels are dissociation curve, profound ascorbate and that there was also a significant re- modified by several mechanisms and may deficiency, and clinical features of scurvy; duction in plasma ascorbate levels. Hap- play a role in both adaptation to anemia these abnormalities were corrected by toglobin levels were significantly re- and vascular damage, there is a strong treatment with vitamin C. -

Iron Deficiency and the Anemia of Chronic Disease
Thomas G. DeLoughery, MD MACP FAWM Professor of Medicine, Pathology, and Pediatrics Oregon Health Sciences University Portland, Oregon [email protected] IRON DEFICIENCY AND THE ANEMIA OF CHRONIC DISEASE SIGNIFICANCE Lack of iron and the anemia of chronic disease are the most common causes of anemia in the world. The majority of pre-menopausal women will have some element of iron deficiency. The first clue to many GI cancers and other diseases is iron loss. Finally, iron deficiency is one of the most treatable medical disorders of the elderly. IRON METABOLISM It is crucial to understand normal iron metabolism to understand iron deficiency and the anemia of chronic disease. Iron in food is largely in ferric form (Fe+++ ) which is reduced by stomach acid to the ferrous form (Fe++). In the jejunum two receptors on the mucosal cells absorb iron. The one for heme-iron (heme iron receptor) is very avid for heme-bound iron (absorbs 30-40%). The other receptor - divalent metal transporter (DMT1) - takes up inorganic iron but is less efficient (1-10%). Iron is exported from the enterocyte via ferroportin and is then delivered to the transferrin receptor (TfR) and then to plasma transferrin. Transferrin is the main transport molecule for iron. Transferrin can deliver iron to the marrow for the use in RBC production or to the liver for storage in ferritin. Transferrin binds to the TfR on the cell and iron is delivered either for use in hemoglobin synthesis or storage. Iron that is contained in hemoglobin in senescent red cells is recycled by binding to ferritin in the macrophage and is transferred to transferrin for recycling. -
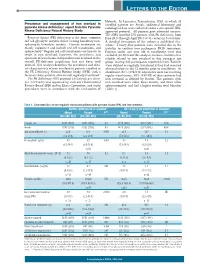
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin. -

The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2
CASE REPORT The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2 1Department of Medicine, Saint Mary Hospital, Nakhon Ratchasima 30000, Thailand, 2Department of General Practice, NopparatRajathanee Hospital, Khanna Yao, Bangkok 10230, Thailand ABSTRACT Polycythemia vera (PV) is a clonal myeloproliferative neoplasm mainly characterized by an abnormal increase of erythroid precursor cells leading to increased red blood cells (RBC) production that is opposite to iron deficiency anemia (IDA) of which the RBC production is decreased due to iron deficiency. This report was aimed to present one patient who had coexistence of these two opposite entities of the RBC production. She was a 47-year-old Thai who was admitted because of acute coronary syndrome and she was accidentally found to have microcytosis of RBC despite normal hemoglobin (Hb) concentration, Hb 14.7 g%, mean corpuscular volume (MCV) 70.0 fL, white blood cells 12,400/mm3, and platelet 401,000/mm3. The Hb analysis showed only A2A, with normal Hb A2 percentage. The polymerase chain reaction for alpha thalassemia-1 genotype was tested negative. Due to neither alpha- nor beta-thalassemia trait detected, the iron study was performed: Serum ferritin 6.1 ng/mL, serum iron 64 ug/dl, and total iron binding capacity 198 ug/dl. The iron storage was seemingly insufficient; hence, iron supplement was started and continued for 4 months. Her blood tests showed: Hb 18.3 g%, MCV 87.2 fl, serum ferritin 31.7 ng/ml, erythropoietin <1 IU/l, positive JAK2 V617F mutation, and normal oxygen saturation. The diagnosis of PV was definitely concluded and she was finally treated with hydroxyurea and occasional phlebotomy. -

Sickle Beta Plus Thalassemia Disease
Sickle Beta Plus Thalassemia Disease WHAT IS SICKLE BETA PLUS THALASSEMIA (Hb Sβ+ thal) Sickle Beta + Thalassemia (Sickle BA-ta Plus Thal-a-SEE-me-a) is a "mild" form of sickle cell disease. Your child's red blood cells contain an abnormal hemoglobin called hemoglobin S or sickle hemoglobin in addition to a small amount of the normal hemoglobin called hemoglobin A. The red blood cells have a defect called beta plus thalassemia, which results in cells which are small in size and more pale than usual. Instead of appearing round or donut shaped, your child's red blood cells are somewhat small, pale, and misshapen. Some may appear sickled or banana shaped. Because sickle beta plus thalassemia is inherited, it is a lifelong disorder. There is no treatment or cure. Your child will always have a mild anemia or slightly low blood count. This may cause occasional tiredness or weakness. HOW DID MY CHILD GET SICKLE BETA PLUS THALASSEMIA When one parent has Sickle Trait (AS) and the other parent has Beta Thalassemia Plus Trait (AT+), them is a l-in-4 chance (25 percent) the baby will have normal hemoglobin (AA), Sickle Trait (AS), Beta Thalassemia Plus Trait (AT+), or Sickle Beta Plus Thalassemia (ST +). These chances remain the same for each pregnancy. PROBLEMS SEEN IN CHILDREN WITH SICKLE BETA PLUS THALASSEMIA Painful episodes can occur with sickle-thalassemia. The sickled red blood cells in sickle-thalassemia, somewhat like those in sickle cell anemia, are rigid and stiff and may sometimes cause "log jams" in the small blood vessels in the bones, organs, and other parts of the body.