Phenotypic and Molecular Genetic Analysis of Pyruvate Kinase Deficiency in a Tunisian Family
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non-Commercial Use Only
only use Non-commercial 14th International Conference on Thalassaemia and Other Haemoglobinopathies 16th TIF Conference for Patients and Parents 17-19 November 2017 • Grand Hotel Palace, Thessaloniki, Greece only use For thalassemia patients with chronic transfusional iron overload... Make a lasting impression with EXJADENon-commercial film-coated tablets The efficacy of deferasirox in a convenient once-daily film-coated tablet Please see your local Novartis representative for Full Product Information Reference: EXJADE® film-coated tablets [EU Summary of Product Characteristics]. Novartis; August 2017. Important note: Before prescribing, consult full prescribing information. iron after having achieved a satisfactory body iron level and therefore retreatment cannot be recommended. ♦ Maximum daily dose is 14 mg/kg body weight. ♦ In pediatric patients the Presentation: Dispersible tablets containing 125 mg, 250 mg or 500 mg of deferasirox. dosing should not exceed 7 mg/kg; closer monitoring of LIC and serum ferritin is essential Film-coated tablets containing 90 mg, 180 mg or 360 mg of deferasirox. to avoid overchelation; in addition to monthly serum ferritin assessments, LIC should be Indications: For the treatment of chronic iron overload due to frequent blood transfusions monitored every 3 months when serum ferritin is ≤800 micrograms/l. (≥7 ml/kg/month of packed red blood cells) in patients with beta-thalassemia major aged Dosage: Special population ♦ In moderate hepatic impairment (Child-Pugh B) dose should 6 years and older. ♦ Also indicated for the treatment of chronic iron overload due to blood not exceed 50% of the normal dose. Should not be used in severe hepatic impairment transfusions when deferoxamine therapy is contraindicated or inadequate in the following (Child-Pugh C). -

Reduced Expression of Pyruvate Kinase in Kidney Proximal Tubule
www.nature.com/scientificreports OPEN Reduced expression of pyruvate kinase in kidney proximal tubule cells is a potential mechanism Received: 29 March 2018 Accepted: 22 January 2019 of pravastatin altered glucose Published: xx xx xxxx metabolism Yong Pyo Lee1, Yuri Cho2, Eun Jee Kim2,3, Hyojung Lee2, Hoon Young Choi4, Hye Jin Wang5, Eun Seok Kang 5, Yu Seun Kim2,6, Myoung Soo Kim1,2,6 & Beom Seok Kim2,3,7 Recent studies have reported that statins are associated with increased incidence of diabetes. Although several mechanisms have been proposed, the role of the kidney’s glucose metabolism upon statin treatment is still unclear. Thus, we investigated the role of pravastatin in gluconeogenesis and glycolysis. HK-2 and HepG2 cells were treated with pravastatin and cultured under either high- or normal-cholesterol conditions. In HK-2 cells treated with pravastatin under both high- and normal- cholesterol conditions, the protein expression of only pyruvate kinase isozymes L/R (PKLR) decreased in a dose-dependent manner, while the protein expression of other glucose metabolism related enzymes remained unchanged. Within the in vivo experiment, male C57BL/6 mice were fed either pravastatin-treated normal-fat diets for 2 or 4 weeks or pravastatin-treated high-fat diets for 16 weeks. Protein expression of PKLR in the kidneys from mice that consumed pravastatin-treated high-fat diets decreased signifcantly compared to the controls. Upon the treatments of pravastatin, only the PKLR expression decreased in lean mice. Furthermore, PKLR activity decreased signifcantly in the kidney after pravastatin treatments. However, there was no change in enzyme activity in the liver, suggesting that pravastatin decreased PKLR activity only in the kidney. -

Hb S/Beta-Thalassemia
rev bras hematol hemoter. 2 0 1 5;3 7(3):150–152 Revista Brasileira de Hematologia e Hemoterapia Brazilian Journal of Hematology and Hemotherapy www.rbhh.org Scientific Comment ଝ The compound state: Hb S/beta-thalassemia ∗ Maria Stella Figueiredo Universidade Federal de São Paulo (UNIFESP), São Paulo, SP, Brazil Sickle cell disease (SCD) results from a single amino acid small increase in Hb concentration and in packed cell volume. substitution in the gene encoding the -globin subunit However, these effects are not accompanied by any reduction  ( 6Glu > Val) that produces the abnormal hemoglobin (Hb) in vaso-occlusive events, probably due to the great number of named Hb S. SCD has different genotypes with substantial Hb S-containing red blood cells resulting in increased blood 1,2 4,5 variations in presentation and clinical course (Table 1). The viscosity. combination of the sickle cell mutation and beta-thalassemia A confusing diagnostic problem is the differentiation 0  (-Thal) mutation gives rise to a compound heterozygous con- of Hb S/ -Thal from sickle cell anemia associated with ␣ dition known as Hb S/ thalassemia (Hb S/-Thal), which was -thalassemia. Table 2 shows that hematologic and elec- 3 first described in 1944 by Silvestroni and Bianco. trophoretic studies are unable to distinguish between the two The polymerization of deoxygenated Hb S (sickling) is the conditions and so family studies and DNA analysis are needed 5 primary event in the molecular pathogenesis of SCD. How- to confirm the diagnosis. +  ever, this event is highly dependent on the intracellular Hb In Hb S/ -Thal, variable amounts of Hb A dilute Hb S and composition; in other words, it is dependent on the concen- consequently inhibit polymerization-induced cellular dam- tration of Hb S, and type and concentration of the other types age. -

Non-Transfusion-Dependent Thalassemias
REVIEW ARTICLES Non-transfusion-dependent thalassemias Khaled M. Musallam, 1 Stefano Rivella, 2 Elliott Vichinsky, 3 and Eliezer A. Rachmilewitz, 4 1Department of Medicine and Medical Specialties, IRCCS Ca’ Granda Foundation Maggiore Policlinico Hospital, University of Milan, Milan, Italy; 2Department of Pediatrics, Division of Hematology/Oncology, Weill Medical College of Cornell University, New York, NY, USA; 3Department of Hematology and Oncology, Children’s Hospital and Research Center Oakland, Oakland, CA, USA; and 4Department of Hematology, Wolfson Medical Center, Holon, Israel ABSTRACT Non-transfusion-dependent thalassemias include a variety of phenotypes that, unlike patients with beta ( β)-tha - lassemia major, do not require regular transfusion therapy for survival. The most commonly investigated forms are β-thalassemia intermedia, hemoglobin E/ β-thalassemia, and α-thalassemia intermedia (hemoglobin H disease). However, transfusion-independence in such patients is not without side effects. Ineffective erythropoiesis and peripheral hemolysis, the hallmarks of disease process, lead to a variety of subsequent pathophysiologies including iron overload and hypercoagulability that ultimately lead to a number of serious clinical morbidities. Thus, prompt and accurate diagnosis of non-transfusion-dependent thalassemia is essential to ensure early intervention. Although several management options are currently available, the need to develop more novel therapeutics is jus - tified by recent advances in our understanding of the mechanisms of disease. Such efforts require wide interna - tional collaboration, especially since non-transfusion-dependent thalassemias are no longer bound to low- and middle-income countries but have spread to large multiethnic cities in Europe and the Americas due to continued migration. Introduction survival, although they may require occasional or even fre - quent transfusions in certain clinical settings and usually for Inherited hemoglobin disorders can be divided into two defined periods of time (Figure 1). -

Supporting Information
Supporting Information Wakabayashi et al. 10.1073/pnas.1521754113 SI Materials and Methods DNA Transfection and Puromycin Selection. K562 cells were cotrans- Plasmid Preparation. Short guide RNA (sgRNA) sequences (Table fected with 1 μg total of Cas9 nuclease and sgRNA plasmids using S1) were cloned into the pSg1 vector (Addgene) and the XPR5 Lipofectamine LTX Plus Reagent (Thermo Fisher Scientific) at a lentiviral vector (Broad Institute), respectively. [The XPR5 1:2 ratio of Cas9 to sgRNA. For a control, K562 cells were co- vector contains the Cas9 nuclease and a red fluorescent protein transfectedwith1μg total of Cas9 nuclease and pLKO.1-GFP (RFP) cassette.] The Cas9 nuclease expression vector used was plasmid at a 1:2 ratio of Cas9 to pLKO.1-GFP. At 24 h after co- μ pxPR_BRD001, which contains a puromycin resistance cassette transfection, puromycin was added at a concentration of 2 g/mL, μ as a selection marker. Off-target scores for each guide were followed 24 h later by a reduction to 1 g/mL for an additional 24 h. calculated using the CRISPR design tool (CRISPR Design; Selection efficiency was assessed by flow cytometry with propidium crispr.mit.edu); only guides with a score >50 (except for a score iodide staining (to assess viability) on a FACSCanto II flow cy- of 49 in one case) were used. tometer (BD Biosciences). Limiting dilutions were performed to obtain single cell-derived clonal populations for both cells targeted Cell Culture and Lentivirus Production. The K562 cells (American with sgRNAs as well as for GFP controls. Unless specified other- Type Culture Collection) were maintained in RPMI medium 1640 wise, three matching clonal GFP controls were analyzed for each plus L-glutamine (Life Technologies) supplemented with 10% experiment. -

The Variable Manifestations of Disease in Pyruvate Kinase Deficiency and Their Management
REVIEW ARTICLE The variable manifestations of disease in pyruvate kinase deficiency and their Ferrata Storti Foundation management Hanny Al-Samkari,1 Eduard J. van Beers,2 Kevin H.M. Kuo,3 Wilma Barcellini,4 Paola Bianchi,4 Andreas Glenthøj,5 María del Mar Mañú Pereira,6 Richard van Wijk,7 Bertil Glader8 and Rachael F. Grace9 1Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; 2Van Creveldkliniek, University Medical Centre Utrecht, University of Utrecht, Utrecht, the Netherlands; 3Division of Hematology, University of Toronto, University Health Haematologica 2020 Network, Toronto, Ontario, Canada; 4UOS Ematologia, Fisiopatologia delle Anemie, Volume 105(9):2229-2239 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy; 5Department of Hematology, Herlev and Gentofte Hospital, Herlev, Denmark; 6Translational Research in Rare Anaemia Disorders, Vall d'Hebron Institut de Recerca (VHIR), Barcelona, Spain; 7Department of Clinical Chemistry & Hematology, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands; 8Lucile Packard Children’s Hospital, Stanford University School of Medicine, Palo Alto, CA, USA and 9Dana/Farber Boston Children's Cancer and Blood Disorders Center, Harvard Medical School, Boston, MA, USA. ABSTRACT yruvate kinase deficiency (PKD) is the most common cause of chron- ic hereditary non-spherocytic hemolytic anemia and results in a Pbroad spectrum of disease. The diagnosis of PKD requires a high index of suspicion and judicious use of laboratory tests that may not always be informative, including pyruvate kinase enzyme assay and genetic analysis of the PKLR gene. A significant minority of patients with PKD have occult mutations in non-coding regions of PKLR which are missed on standard genetic tests. -

Protein Interactions in the Cancer Proteome† Cite This: Mol
Molecular BioSystems View Article Online PAPER View Journal | View Issue Small-molecule binding sites to explore protein– protein interactions in the cancer proteome† Cite this: Mol. BioSyst., 2016, 12,3067 David Xu,ab Shadia I. Jalal,c George W. Sledge Jr.d and Samy O. Meroueh*aef The Cancer Genome Atlas (TCGA) offers an unprecedented opportunity to identify small-molecule binding sites on proteins with overexpressed mRNA levels that correlate with poor survival. Here, we analyze RNA-seq and clinical data for 10 tumor types to identify genes that are both overexpressed and correlate with patient survival. Protein products of these genes were scanned for binding sites that possess shape and physicochemical properties that can accommodate small-molecule probes or therapeutic agents (druggable). These binding sites were classified as enzyme active sites (ENZ), protein–protein interaction sites (PPI), or other sites whose function is unknown (OTH). Interestingly, the overwhelming majority of binding sites were classified as OTH. We find that ENZ, PPI, and OTH binding sites often occurred on the same structure suggesting that many of these OTH cavities can be used for allosteric modulation of Creative Commons Attribution 3.0 Unported Licence. enzyme activity or protein–protein interactions with small molecules. We discovered several ENZ (PYCR1, QPRT,andHSPA6)andPPI(CASC5, ZBTB32,andCSAD) binding sites on proteins that have been seldom explored in cancer. We also found proteins that have been extensively studied in cancer that have not been previously explored with small molecules that harbor ENZ (PKMYT1, STEAP3,andNNMT) and PPI (HNF4A, MEF2B,andCBX2) binding sites. All binding sites were classified by the signaling pathways to Received 29th March 2016, which the protein that harbors them belongs using KEGG. -

Chrebp Regulates Fructose-Induced Glucose Production Independently of Insulin Signaling
ChREBP regulates fructose-induced glucose production independently of insulin signaling Mi-Sung Kim, … , Michelle Lai, Mark A. Herman J Clin Invest. 2016;126(11):4372-4386. https://doi.org/10.1172/JCI81993. Research Article Endocrinology Metabolism Obese, insulin-resistant states are characterized by a paradoxical pathogenic condition in which the liver appears to be selectively insulin resistant. Specifically, insulin fails to suppress glucose production, yet successfully stimulates de novo lipogenesis. The mechanisms underlying this dysregulation remain controversial. Here, we hypothesized that carbohydrate-responsive element-binding protein (ChREBP), a transcriptional activator of glycolytic and lipogenic genes, plays a central role in this paradox. Administration of fructose increased hepatic hexose-phosphate levels, activated ChREBP, and caused glucose intolerance, hyperinsulinemia, hypertriglyceridemia, and hepatic steatosis in mice. Activation of ChREBP was required for the increased expression of glycolytic and lipogenic genes as well as glucose-6- phosphatase (G6pc) that was associated with the effects of fructose administration. We found that fructose-induced G6PC activity is a major determinant of hepatic glucose production and reduces hepatic glucose-6-phosphate levels to complete a homeostatic loop. Moreover, fructose activated ChREBP and induced G6pc in the absence of Foxo1a, indicating that carbohydrate-induced activation of ChREBP and G6PC dominates over the suppressive effects of insulin to enhance glucose production. This ChREBP/G6PC signaling axis is conserved in humans. Together, these findings support a carbohydrate-mediated, ChREBP-driven mechanism that contributes to hepatic insulin resistance. Find the latest version: https://jci.me/81993/pdf RESEARCH ARTICLE The Journal of Clinical Investigation ChREBP regulates fructose-induced glucose production independently of insulin signaling Mi-Sung Kim,1 Sarah A. -

Glucose Catabolism in Liver Tumors Induced by C-MYC Can Be
Published OnlineFirst June 19, 2017; DOI: 10.1158/0008-5472.CAN-17-0498 Cancer Tumor and Stem Cell Biology Research Glucose Catabolism in Liver Tumors Induced by c-MYC Can Be Sustained by Various PKM1/PKM2 Ratios and Pyruvate Kinase Activities Andres Mendez-Lucas 1, Xiaolei Li2,3, Junjie Hu2,4, Li Che2, Xinhua Song2, Jiaoyuan Jia2,5, Jingxiao Wang2, Chencheng Xie2,6, Paul C. Driscoll1, Darjus F. Tschaharganeh7,8, Diego F. Calvisi9, Mariia Yuneva1, and Xin Chen2,4 Abstract Different pyruvate kinase isoforms are expressed in a tissue- tumorigenesis and resulted in liver tumor formation with specific manner, with pyruvate kinase M2 (PKM2) suggested to decreased pyruvate kinase activity and decreased catabolism be the predominant isoform in proliferating cells and cancer of glucose into alanine and the Krebs cycle. An increased PKM1/ cells. Because of differential regulation of enzymatic activities, PKM2 ratio by ectopic PKM1 expression further decreased PKM2, but not PKM1, has been thought to favor cell prolifer- glucose flux into serine biosynthesis and increased flux into ation. However, the role of PKM2 in tumorigenesis has been lactate and the Krebs cycle, resulting in reduced total levels of recently challenged. Here we report that increased glucose serine. However, these changes also did not affect c-MYC– catabolism through glycolysis and increased pyruvate kinase induced liver tumor development. These results suggest that activity in c-MYC-driven liver tumors are associated with increased expression of PKM2 is not required to support increased expression of both PKM1 and PKM2 isoforms and c-MYC–induced tumorigenesis in the liver and that various decreased expression of the liver-specific isoform of pyruvate PKM1/PKM2 ratios and pyruvate kinase activities can sustain kinase, PKL. -

Chrebp-Knockout Mice Show Sucrose Intolerance and Fructose Malabsorption
nutrients Article ChREBP-Knockout Mice Show Sucrose Intolerance and Fructose Malabsorption Takehiro Kato 1, Katsumi Iizuka 1,2,* ID , Ken Takao 1, Yukio Horikawa 1, Tadahiro Kitamura 3 and Jun Takeda 1 1 Department of Diabetes and Endocrinology, Graduate School of Medicine, Gifu University, Gifu 501-1194, Japan; [email protected] (T.K.); [email protected] (K.T.); [email protected] (Y.H.); [email protected] (J.T.) 2 Gifu University Hospital Center for Nutritional Support and Infection Control, Gifu 501-1194, Japan 3 Metabolic Signal Research Center, Institute for Molecular and Cellular Regulation, Gunma University, Gunma 371-8512, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-58-230-6564; Fax: +81-58-230-6376 Received: 31 January 2018; Accepted: 9 March 2018; Published: 10 March 2018 Abstract: We have previously reported that 60% sucrose diet-fed ChREBP knockout mice (KO) showed body weight loss resulting in lethality. We aimed to elucidate whether sucrose and fructose metabolism are impaired in KO. Wild-type mice (WT) and KO were fed a diet containing 30% sucrose with/without 0.08% miglitol, an α-glucosidase inhibitor, and these effects on phenotypes were tested. Furthermore, we compared metabolic changes of oral and peritoneal fructose injection. A thirty percent sucrose diet feeding did not affect phenotypes in KO. However, miglitol induced lethality in 30% sucrose-fed KO. Thirty percent sucrose plus miglitol diet-fed KO showed increased cecal contents, increased fecal lactate contents, increased growth of lactobacillales and Bifidobacterium and decreased growth of clostridium cluster XIVa. -

Iron Deficiency and the Anemia of Chronic Disease
Thomas G. DeLoughery, MD MACP FAWM Professor of Medicine, Pathology, and Pediatrics Oregon Health Sciences University Portland, Oregon [email protected] IRON DEFICIENCY AND THE ANEMIA OF CHRONIC DISEASE SIGNIFICANCE Lack of iron and the anemia of chronic disease are the most common causes of anemia in the world. The majority of pre-menopausal women will have some element of iron deficiency. The first clue to many GI cancers and other diseases is iron loss. Finally, iron deficiency is one of the most treatable medical disorders of the elderly. IRON METABOLISM It is crucial to understand normal iron metabolism to understand iron deficiency and the anemia of chronic disease. Iron in food is largely in ferric form (Fe+++ ) which is reduced by stomach acid to the ferrous form (Fe++). In the jejunum two receptors on the mucosal cells absorb iron. The one for heme-iron (heme iron receptor) is very avid for heme-bound iron (absorbs 30-40%). The other receptor - divalent metal transporter (DMT1) - takes up inorganic iron but is less efficient (1-10%). Iron is exported from the enterocyte via ferroportin and is then delivered to the transferrin receptor (TfR) and then to plasma transferrin. Transferrin is the main transport molecule for iron. Transferrin can deliver iron to the marrow for the use in RBC production or to the liver for storage in ferritin. Transferrin binds to the TfR on the cell and iron is delivered either for use in hemoglobin synthesis or storage. Iron that is contained in hemoglobin in senescent red cells is recycled by binding to ferritin in the macrophage and is transferred to transferrin for recycling. -
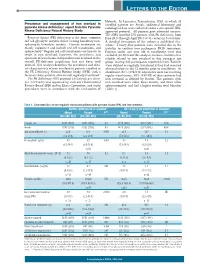
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin.