Texas DSHS Handout Alpha Thalassemia Trait
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non-Commercial Use Only
only use Non-commercial 14th International Conference on Thalassaemia and Other Haemoglobinopathies 16th TIF Conference for Patients and Parents 17-19 November 2017 • Grand Hotel Palace, Thessaloniki, Greece only use For thalassemia patients with chronic transfusional iron overload... Make a lasting impression with EXJADENon-commercial film-coated tablets The efficacy of deferasirox in a convenient once-daily film-coated tablet Please see your local Novartis representative for Full Product Information Reference: EXJADE® film-coated tablets [EU Summary of Product Characteristics]. Novartis; August 2017. Important note: Before prescribing, consult full prescribing information. iron after having achieved a satisfactory body iron level and therefore retreatment cannot be recommended. ♦ Maximum daily dose is 14 mg/kg body weight. ♦ In pediatric patients the Presentation: Dispersible tablets containing 125 mg, 250 mg or 500 mg of deferasirox. dosing should not exceed 7 mg/kg; closer monitoring of LIC and serum ferritin is essential Film-coated tablets containing 90 mg, 180 mg or 360 mg of deferasirox. to avoid overchelation; in addition to monthly serum ferritin assessments, LIC should be Indications: For the treatment of chronic iron overload due to frequent blood transfusions monitored every 3 months when serum ferritin is ≤800 micrograms/l. (≥7 ml/kg/month of packed red blood cells) in patients with beta-thalassemia major aged Dosage: Special population ♦ In moderate hepatic impairment (Child-Pugh B) dose should 6 years and older. ♦ Also indicated for the treatment of chronic iron overload due to blood not exceed 50% of the normal dose. Should not be used in severe hepatic impairment transfusions when deferoxamine therapy is contraindicated or inadequate in the following (Child-Pugh C). -

Hereditary Spherocytosis: Clinical Features
Title Overview: Hereditary Hematological Disorders of red cell shape. Disorders Red cell Enzyme disorders Disorders of Hemoglobin Inherited bleeding disorders- platelet disorders, coagulation factor Anthea Greenway MBBS FRACP FRCPA Visiting Associate deficiencies Division of Pediatric Hematology-Oncology Duke University Health Service Inherited Thrombophilia Hereditary Disorders of red cell Disorders of red cell shape (cytoskeleton): cytoskeleton: • Mutations of 5 proteins connect cytoskeleton of red cell to red cell membrane • Hereditary Spherocytosis- sphere – Spectrin (composed of alpha, beta heterodimers) –Ankyrin • Hereditary Elliptocytosis-ellipse, elongated forms – Pallidin (band 4.2) – Band 4.1 (protein 4.1) • Hereditary Pyropoikilocytosis-bizarre red cell forms – Band 3 protein (the anion exchanger, AE1) – RhAG (the Rh-associated glycoprotein) Normal red blood cell- discoid, with membrane flexibility Hereditary Spherocytosis: Clinical features: • Most common hereditary hemolytic disorder (red cell • Neonatal jaundice- severe (phototherapy), +/- anaemia membrane) • Hemolytic anemia- moderate in 60-75% cases • Mutations of one of 5 genes (chromosome 8) for • Severe hemolytic anaemia in 5% (AR, parents ASx) cytoskeletal proteins, overall effect is spectrin • fatigue, jaundice, dark urine deficiency, severity dependant on spectrin deficiency • SplenomegalSplenomegaly • 200-300:million births, most common in Northern • Chronic complications- growth impairment, gallstones European countries • Often follows clinical course of affected -

Newborn Screening Result for Bart's Hemoglobin
NEWBORN SCREENING RESULT FOR BART’S HEMOGLOBIN Physician’s information sheet developed by the Nebraska Newborn Screening Program with review by James Harper, MD, Pediatric Hematologist with UNMC Follow-up program, and member of the Nebraska Newborn Screening Advisory Committee. BACKGROUND The alpha thalassemias result from the loss of alpha globin genes. There are normally four genes for alpha globin production so that the loss of one to four genes is possible. The lack of one gene causes alpha thalassemia 2 (silent carrier) with no clinically detectable problems but may cause small amounts of hemoglobin Barts to be present in newborn blood samples. Alpha thalassemia trait (Alpha thalassemia 1) results from loss of two genes and causes a mild microcytic anemia which may resemble iron deficiency anemia. The loss of three genes causes hemoglobin H diseases which is a moderately severe form of thalassemia. The lack of all four genes causes hydrops fetalis and is usually fatal in utero. In general, only the loss of one or two genes is seen in African Americans. Individuals from Southeast Asia and the Mediterranean may have all four types of alpha thalassemia. The percentage of hemoglobin Barts in the blood sample may indicate the number of alpha genes that have been lost. However, the percentage of hemoglobin Barts is not directly measurable with the current methodology used by the newborn screening laboratory. Only the presence of Barts hemoglobin in relation to fetal and adult hemoglobin, and variants S, C, D and E can be detected. RECOMMENDED WORK UP In addition to the standard newborn hemoglobinopathy confirmation (hemoglobin electrophoresis), to separate those patients with alpha thalassemia silent carrier from the patients with alpha thalassemia trait, we recommend that these babies have the following labs drawn at their 6 month well baby check: CBC with retic count, ferritin, and a hemoglobin electropheresis. -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -

Methemoglobinemia and Ascorbate Deficiency in Hemoglobin E Β Thalassemia: Metabolic and Clinical Implications
From www.bloodjournal.org by guest on April 2, 2015. For personal use only. Plenary paper Methemoglobinemia and ascorbate deficiency in hemoglobin E  thalassemia: metabolic and clinical implications Angela Allen,1,2 Christopher Fisher,1 Anuja Premawardhena,3 Dayananda Bandara,4 Ashok Perera,4 Stephen Allen,2 Timothy St Pierre,5 Nancy Olivieri,6 and David Weatherall1 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom; 2College of Medicine, Swansea University, Swansea, United Kingdom; 3University of Kelaniya, Colombo, Sri Lanka; 4National Thalassaemia Centre, District Hospital, Kurunegala, Sri Lanka; 5School of Physics, University of Western Australia, Crawley, Australia; and 6Hemoglobinopathy Research, University Health Network, Toronto, ON During investigations of the phenotypic man hypoxia induction factor pathway is There was, in addition, a highly signifi- diversity of hemoglobin (Hb) E  thalasse- not totally dependent on ascorbate lev- cant correlation between methemoglobin mia, a patient was encountered with per- els. A follow-up study of 45 patients with levels, splenectomy, and factors that sistently high levels of methemoglobin HbE  thalassemia showed that methemo- modify the degree of globin-chain imbal- associated with a left-shift in the oxygen globin levels were significantly increased ance. Because methemoglobin levels are dissociation curve, profound ascorbate and that there was also a significant re- modified by several mechanisms and may deficiency, and clinical features of scurvy; duction in plasma ascorbate levels. Hap- play a role in both adaptation to anemia these abnormalities were corrected by toglobin levels were significantly re- and vascular damage, there is a strong treatment with vitamin C. -

Acoi Board Review 2019 Text
CHERYL KOVALSKI, DO FACOI NO DISCLOSURES ACOI BOARD REVIEW 2019 TEXT ANEMIA ‣ Hemoglobin <13 grams or ‣ Hematocrit<39% TEXT ANEMIA MCV RETICULOCYTE COUNT Corrected retic ct = hematocrit/45 x retic % (45 considered normal hematocrit) >2%: blood loss or hemolysis <2%: hypoproliferative process TEXT ANEMIA ‣ MICROCYTIC ‣ Obtain and interpret iron studies ‣ Serum iron ‣ Total iron binding capacity (TIBC) ‣ Transferrin saturation ‣ Ferritin-correlates with total iron stores ‣ can be normal or increased if co-existent inflammation TEXT IRON DEFICIENCY ‣ Most common nutritional problem in the world ‣ Absorbed in small bowel, enhanced by gastric acid ‣ Absorption inhibited by inflammation, phytates (bran) & tannins (tea) TEXT CAUSES OF IRON DEFICIENCY ‣ Blood loss – most common etiology ‣ Decreased intake ‣ Increased utilization-EPO therapy, chronic hemolysis ‣ Malabsorption – gastrectomy, sprue ‣ ‣ ‣ TEXT CLINICAL MANIFESTATIONS OF IRON DEFICIENCY ‣ Impaired psychomotor development ‣ Fatigue, Irritability ‣ PICA ‣ Koilonychiae, Glossitis, Angular stomatitis ‣ Dysphagia TEXT IRON DEFICIENCY LAB FINDINGS ‣ Low serum iron, increased TIBC ‣ % sat <20 TEXT MANAGEMENT OF IRON DEFICIENCY ‣ MUST LOOK FOR SOURCE OF BLEED: ie: GI, GU, Regular blood donor ‣ Replacement: 1. Oral: Ferrous sulfate 325 mg TID until serum iron, % sat, and ferritin mid-range normal, 6-12 months 2. IV TEXT SIDEROBLASTIC ANEMIAS Diverse group of disorders of RBC production characterized by: 1. Defect involving incorporation of iron into heme molecule 2. Ringed sideroblasts in -

Approach to Anemia
APPROACH TO ANEMIA Mahsa Mohebtash, MD Medstar Union Memorial Hospital Definition of Anemia • Reduced red blood mass • RBC measurements: RBC mass, Hgb, Hct or RBC count • Hgb, Hct and RBC count typically decrease in parallel except in severe microcytosis (like thalassemia) Normal Range of Hgb/Hct • NL range: many different values: • 2 SD below mean: < Hgb13.5 or Hct 41 in men and Hgb 12 or Hct of 36 in women • WHO: Hgb: <13 in men, <12 in women • Revised WHO/NCI: Hgb <14 in men, <12 in women • Scrpps-Kaiser based on race and age: based on 5th percentiles of the population in question • African-Americans: Hgb 0.5-1 lower than Caucasians Approach to Anemia • Setting: • Acute vs chronic • Isolated vs combined with leukopenia/thrombocytopenia • Pathophysiologic approach • Morphologic approach Reticulocytes • Reticulocytes life span: 3 days in bone marrow and 1 day in peripheral blood • Mature RBC life span: 110-120 days • 1% of RBCs are removed from circulation each day • Reticulocyte production index (RPI): Reticulocytes (percent) x (HCT ÷ 45) x (1 ÷ RMT): • <2 low Pathophysiologic approach • Decreased RBC production • Reduced effective production of red cells: low retic production index • Destruction of red cell precursors in marrow (ineffective erythropoiesis) • Increased RBC destruction • Blood loss Reduced RBC precursors • Low retic production index • Lack of nutrients (B12, Fe) • Bone marrow disorder => reduced RBC precursors (aplastic anemia, pure RBC aplasia, marrow infiltration) • Bone marrow suppression (drugs, chemotherapy, radiation) -

Clinical Evaluation of Different Types of Anemia 1Richa Saxena, 2Sudha Chamoli, 3Monisha Batra
WJOA Richa Saxena et al 10.5005/jp-journals-10065-0024 REVIEW ARTICLE Clinical Evaluation of Different Types of Anemia 1Richa Saxena, 2Sudha Chamoli, 3Monisha Batra World Health Organization criteria for anemia ABSTRACT Table 1: Venous blood (gm/dL) MCHC Anemia, defined as a hemoglobin level two standard devia- Adult males 13 34 tions below the mean for age, is prevalent among infants and children as well as adults worldwide. The evaluation Adult females, nonpregnant 12 34 of an individual with anemia should begin with a thorough Adult females, pregnant 11 34 history and risk assessment. Characterizing the anemia as Children (6 months–6 years) 11 34 microcytic, normocytic, or macrocytic based on the mean Children (6–14 years) 12 34 corpuscular volume (MCV) will aid in the work-up and man- agement. Microcytic anemia due to iron deficiency is the most common type of anemia in children. Iron deficiency CLASSIFICATION OF ANEMIA anemia, which can be associated with cognitive issues, is prevented and treated with iron supplements or increased The classification of anemia based on two factors (Table 2): intake of dietary iron. This review article discusses the clinical 1. Red cell morphology evaluation of different types of anemias based on the find- 2. Etiology of anemia ings of clinical examination (i.e., pallor, pedal edema, nail changes, and epithelial changes) as well as the results of Anemia Classification Based on Morphology various investigations such as routine blood investigations (hemoglobin, mean cell hemoglobin concentration [MCHC], Anemia can be classified based on morphology as: packed cell volume, etc.), peripheral smear examination, • Normocytic normochromic (MCV 76–96 fL, MCHC bone marrow examination, etc. -

Iron Deficiency and the Anemia of Chronic Disease
Thomas G. DeLoughery, MD MACP FAWM Professor of Medicine, Pathology, and Pediatrics Oregon Health Sciences University Portland, Oregon [email protected] IRON DEFICIENCY AND THE ANEMIA OF CHRONIC DISEASE SIGNIFICANCE Lack of iron and the anemia of chronic disease are the most common causes of anemia in the world. The majority of pre-menopausal women will have some element of iron deficiency. The first clue to many GI cancers and other diseases is iron loss. Finally, iron deficiency is one of the most treatable medical disorders of the elderly. IRON METABOLISM It is crucial to understand normal iron metabolism to understand iron deficiency and the anemia of chronic disease. Iron in food is largely in ferric form (Fe+++ ) which is reduced by stomach acid to the ferrous form (Fe++). In the jejunum two receptors on the mucosal cells absorb iron. The one for heme-iron (heme iron receptor) is very avid for heme-bound iron (absorbs 30-40%). The other receptor - divalent metal transporter (DMT1) - takes up inorganic iron but is less efficient (1-10%). Iron is exported from the enterocyte via ferroportin and is then delivered to the transferrin receptor (TfR) and then to plasma transferrin. Transferrin is the main transport molecule for iron. Transferrin can deliver iron to the marrow for the use in RBC production or to the liver for storage in ferritin. Transferrin binds to the TfR on the cell and iron is delivered either for use in hemoglobin synthesis or storage. Iron that is contained in hemoglobin in senescent red cells is recycled by binding to ferritin in the macrophage and is transferred to transferrin for recycling. -

Microcytic Hypochromic Anemia Patients with Thalassemia: Genotyping Approach
101 MICROCYTIC HYPOCHROMIC ANEMIA PATIENTS WITH THALASSEMIA: GENOTYPING APPROACH FAKHER RAHIM ABSTRACT BACKGROUND: Microcytic hypochromic anemia is a common condition in clinical practice, and alpha-thalassemia has to be considered as a differential diagnosis. AIMS: This study was conducted to evaluate the frequency of α-gene, β-gene and hemoglobin variant numbers in subjects with microcytic hypochromic anemia. SETTING AND DESIGNS: Population-based case-control study in the Iranian population. MATERIALS AND METHODS: A total of 340 subjects from southwest part of Iran were studied in the Research Center of Thalassemia and Hemoglobinopathies (RCTH), Iran. Genotyping for known α- and β-gene mutations was done with gap-PCR and ARMS. In cases of some rare mutations, the genotyping was done with the help of other techniques such as RFLP and ARMS-PCR. STATISTICAL ANALYSIS: Statistical analysis was carried out by SPSS 11.5 and an independent-sample t test. RESULTS: Out of the total 340 individuals, 325 individuals were evaluated to have microcytic hypochromic anemia based on initial hematological parameters such as MCV<80 fl; MCH <27 pg; the remaining 15 patients were diagnosed with no definite etiology. The overall frequency of -α3.7 deletion in 325 individuals was 20.3%. The most frequent mutations were IVS II-I, CD 36/37 and IVS I-110 with frequencies of 6.31%, 5.27% and 1.64%, respectively. Only, there was a significant difference between beta-thalassemia trait and beta-thalassemia major with regard to MCV (P < 0.05) and MCH (P < 0.05) indices, and also MCH index between beta-thalassemia trait and Hb variants (P < 0.05). -
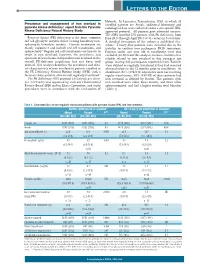
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin. -

The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2
CASE REPORT The Coexistence of Polycythemia Vera and Iron Deficiency Anemia Somchai Insiripong1, Wattana Insiripong2 1Department of Medicine, Saint Mary Hospital, Nakhon Ratchasima 30000, Thailand, 2Department of General Practice, NopparatRajathanee Hospital, Khanna Yao, Bangkok 10230, Thailand ABSTRACT Polycythemia vera (PV) is a clonal myeloproliferative neoplasm mainly characterized by an abnormal increase of erythroid precursor cells leading to increased red blood cells (RBC) production that is opposite to iron deficiency anemia (IDA) of which the RBC production is decreased due to iron deficiency. This report was aimed to present one patient who had coexistence of these two opposite entities of the RBC production. She was a 47-year-old Thai who was admitted because of acute coronary syndrome and she was accidentally found to have microcytosis of RBC despite normal hemoglobin (Hb) concentration, Hb 14.7 g%, mean corpuscular volume (MCV) 70.0 fL, white blood cells 12,400/mm3, and platelet 401,000/mm3. The Hb analysis showed only A2A, with normal Hb A2 percentage. The polymerase chain reaction for alpha thalassemia-1 genotype was tested negative. Due to neither alpha- nor beta-thalassemia trait detected, the iron study was performed: Serum ferritin 6.1 ng/mL, serum iron 64 ug/dl, and total iron binding capacity 198 ug/dl. The iron storage was seemingly insufficient; hence, iron supplement was started and continued for 4 months. Her blood tests showed: Hb 18.3 g%, MCV 87.2 fl, serum ferritin 31.7 ng/ml, erythropoietin <1 IU/l, positive JAK2 V617F mutation, and normal oxygen saturation. The diagnosis of PV was definitely concluded and she was finally treated with hydroxyurea and occasional phlebotomy.