OR51A7 (NM 001004749) Human Untagged Clone – SC300787
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Single Cell Derived Clonal Analysis of Human Glioblastoma Links
SUPPLEMENTARY INFORMATION: Single cell derived clonal analysis of human glioblastoma links functional and genomic heterogeneity ! Mona Meyer*, Jüri Reimand*, Xiaoyang Lan, Renee Head, Xueming Zhu, Michelle Kushida, Jane Bayani, Jessica C. Pressey, Anath Lionel, Ian D. Clarke, Michael Cusimano, Jeremy Squire, Stephen Scherer, Mark Bernstein, Melanie A. Woodin, Gary D. Bader**, and Peter B. Dirks**! ! * These authors contributed equally to this work.! ** Correspondence: [email protected] or [email protected]! ! Supplementary information - Meyer, Reimand et al. Supplementary methods" 4" Patient samples and fluorescence activated cell sorting (FACS)! 4! Differentiation! 4! Immunocytochemistry and EdU Imaging! 4! Proliferation! 5! Western blotting ! 5! Temozolomide treatment! 5! NCI drug library screen! 6! Orthotopic injections! 6! Immunohistochemistry on tumor sections! 6! Promoter methylation of MGMT! 6! Fluorescence in situ Hybridization (FISH)! 7! SNP6 microarray analysis and genome segmentation! 7! Calling copy number alterations! 8! Mapping altered genome segments to genes! 8! Recurrently altered genes with clonal variability! 9! Global analyses of copy number alterations! 9! Phylogenetic analysis of copy number alterations! 10! Microarray analysis! 10! Gene expression differences of TMZ resistant and sensitive clones of GBM-482! 10! Reverse transcription-PCR analyses! 11! Tumor subtype analysis of TMZ-sensitive and resistant clones! 11! Pathway analysis of gene expression in the TMZ-sensitive clone of GBM-482! 11! Supplementary figures and tables" 13" "2 Supplementary information - Meyer, Reimand et al. Table S1: Individual clones from all patient tumors are tumorigenic. ! 14! Fig. S1: clonal tumorigenicity.! 15! Fig. S2: clonal heterogeneity of EGFR and PTEN expression.! 20! Fig. S3: clonal heterogeneity of proliferation.! 21! Fig. -

Identification of Loci Contributing to the Smith- Magenis Syndrome-Like Phenotype and Molecular Evaluation of the Retinoic Acid Induced 1 Gene
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2010 IDENTIFICATION OF LOCI CONTRIBUTING TO THE SMITH- MAGENIS SYNDROME-LIKE PHENOTYPE AND MOLECULAR EVALUATION OF THE RETINOIC ACID INDUCED 1 GENE Stephen Williams Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Medical Genetics Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/65 This Dissertation is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. © Stephen Richardson Williams May 2010 All Rights Reserved ii IDENTIFICATION OF LOCI CONTRIBUTING TO THE SMITH-MAGENIS SYNDROME- LIKE PHENOTYPE AND MOLECULAR EVALUATION OF THE RETINOIC ACID INDUCED 1 GENE A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy at Virginia Commonwealth University. By STEPHEN RICHARDSON WILLIAMS Bachelor of Science (B.S.) James Madison University, Harrisonburg, Virginia, 2001 Director: Sarah H. Elsea, Ph.D., F.A.C.M.G. Associate Professor, Departments of Pediatrics and Human and Molecular Genetics Virginia Commonwealth University Richmond, Virginia May, 2010 iii Acknowledgements There are many people who contributed to my success at VCU both directly and indirectly and I would like to thank those that had the largest impact on my pursuit of scientific knowledge. First and foremost, I thank my mentor Dr. Sarah H. Elsea. Her knowledge and guidance are unmatched and I feel lucky to have work with and under her. -

Genetic Characterization of Greek Population Isolates Reveals Strong Genetic Drift at Missense and Trait-Associated Variants
ARTICLE Received 22 Apr 2014 | Accepted 22 Sep 2014 | Published 6 Nov 2014 DOI: 10.1038/ncomms6345 OPEN Genetic characterization of Greek population isolates reveals strong genetic drift at missense and trait-associated variants Kalliope Panoutsopoulou1,*, Konstantinos Hatzikotoulas1,*, Dionysia Kiara Xifara2,3, Vincenza Colonna4, Aliki-Eleni Farmaki5, Graham R.S. Ritchie1,6, Lorraine Southam1,2, Arthur Gilly1, Ioanna Tachmazidou1, Segun Fatumo1,7,8, Angela Matchan1, Nigel W. Rayner1,2,9, Ioanna Ntalla5,10, Massimo Mezzavilla1,11, Yuan Chen1, Chrysoula Kiagiadaki12, Eleni Zengini13,14, Vasiliki Mamakou13,15, Antonis Athanasiadis16, Margarita Giannakopoulou17, Vassiliki-Eirini Kariakli5, Rebecca N. Nsubuga18, Alex Karabarinde18, Manjinder Sandhu1,8, Gil McVean2, Chris Tyler-Smith1, Emmanouil Tsafantakis12, Maria Karaleftheri16, Yali Xue1, George Dedoussis5 & Eleftheria Zeggini1 Isolated populations are emerging as a powerful study design in the search for low-frequency and rare variant associations with complex phenotypes. Here we genotype 2,296 samples from two isolated Greek populations, the Pomak villages (HELIC-Pomak) in the North of Greece and the Mylopotamos villages (HELIC-MANOLIS) in Crete. We compare their genomic characteristics to the general Greek population and establish them as genetic isolates. In the MANOLIS cohort, we observe an enrichment of missense variants among the variants that have drifted up in frequency by more than fivefold. In the Pomak cohort, we find novel associations at variants on chr11p15.4 showing large allele frequency increases (from 0.2% in the general Greek population to 4.6% in the isolate) with haematological traits, for example, with mean corpuscular volume (rs7116019, P ¼ 2.3 Â 10 À 26). We replicate this association in a second set of Pomak samples (combined P ¼ 2.0 Â 10 À 36). -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -
Exome Array Analysis Identifies GPR35 As a Novel Susceptibility Gene for Anthracycline-Induced Cardiotoxicity in Childhood Cancer
Exome array analysis identifies GPR35 as a novel susceptibility gene for anthracycline-induced cardiotoxicity in childhood cancer Sara Ruiz-Pinto1, Guillermo Pita1, Ana Patiño-García, PhD 2, Purificación García-Miguel, MD3, Javier Alonso, MD4, Antonio Pérez-Martínez, MD, PhD3, Antonio J Cartón, MD, PhD 5, Federico Gutiérrez-Larraya, MD, PhD 5, María R Alonso1, Daniel R. Barnes, PhD6, Joe Dennis7, Kyriaki Michailidou, PhD 6,8, Carmen Gómez-Santos9, Deborah J. Thompson, PhD 7, Douglas F. Easton, PhD 6,7, Javier Benítez, PhD 1,10, Anna González-Neira, PhD 1 1 Human Genotyping Unit-CeGen, Human Cancer Genetics Programme. Spanish National Cancer Research Centre (CNIO), Madrid, 28029, Spain 2 Department of Pediatrics, Universidad de Navarra, University Clinic of Navarra, Pamplona, 31008, Spain 3 Department of Pediatric Hemato-Oncology, Hospital Universitario La Paz, Madrid, 28046, Spain 4 Pediatric solid tumor laboratory. Human Genetic Department. Research Institute of Rare Diseases. Instituto de Salud Carlos III, Majadahonda, 28220, Madrid, Spain 5 Department of Pediatric Cardiology, Hospital Universitario La Paz, Madrid, 28046, Spain 6 Department of Public Health and Primary Care, Centre for Cancer Genetic Epidemiology, Cambridge, CB1 8RN, UK 7 Department of Oncology, Centre for Cancer Genetic Epidemiology, University of Cambridge, Cambridge, CB1 8RN, UK 1 8 Department of Electron Microscopy/Molecular Pathology, Cyprus Institute of Neurology and Genetics, Nicosia, 1683, Cyprus 9 Department of Pediatrics, Hospital Universitario Infanta Elena, 28342, Madrid, Spain 10 Human Genetics Group, Human Cancer Genetics Programme, Spanish National Cancer Research Centre (CNIO), Madrid, 28029, Spain CORRESPONDING AUTHOR INFORMATION Dr Anna González-Neira Human Genotyping Unit-CeGen, Human Cancer Genetics Programme, Spanish National Cancer Centre, Melchor Fernández Almagro 3, Madrid 28029, Spain. -

SUPPLEMENTARY APPENDIX Whole Exome Sequencing Identifies Mutational Signatures of Vitreoretinal Lymphoma
SUPPLEMENTARY APPENDIX Whole exome sequencing identifies mutational signatures of vitreoretinal lymphoma Junwon Lee, 1* Borahm Kim, 2* Hyeonah Lee, 3 Heejung Park, 4 Suk Ho Byeon, 1 Jong Rak Choi, 2 Sung Chul Lee, 1 Seung-Tae Lee 2 and Christopher Seungkyu Lee 1 *JL and BK contributed equally as co-first authors 1Department of Ophthalmology, Severance Hospital, Institute of Vision Research, Yonsei University College of Medicine; 2Department of Laboratory Medi - cine, Yonsei University College of Medicine; 3Brain Korea 21 PLUS Project for Medical Science, Yonsei University and 4Department of pathology, Yonsei Uni - versity College of medicine, Seoul, South Korea Correspondence: CHRISTOPHER SEUNGKYU LEE - [email protected] SEUNG-TAE LEE - [email protected] doi:10.3324/haematol.2019.233783 Supplemental Data Supplemental Methods Supplemental Table 1 Supplemental Figure 1-5 Supplementary Methods Whole exome sequencing (WES) Genomic DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen). The sequencing libraries for Exome‐sequencing were prepared using the Twist Human Core Exome Kit (Twist Bioscience). Paired‐end 100 bp read sequencing was performed on a NovaSeq system (Illumina). The paired‐end reads were mapped to the human genome (NCBI build 37) using BWA (version 0.7.12).1 The alignment was further refined by the functions of local realignment, base quality recalibration and indel realignment provided by GATK software 3.8-0. In order to identify single nucleotide variations (SNVs) and indels, we used HaplotypeCaller and Mutect2 from the GATK package (3.8-0), and VarScan2 (2.4.0). The results of these three algorithms were compared and merged.2-4 An R package, ExomeDepth (version 1.1.10), was used to detect exon- or gene-level copy number variation (CNV) in the target regions,5 followed by visualization using a base-level read depth normalization algorithm implemented in the DxSeq Analyzer (Dxome). -

Mining High-Level Brain Imaging Genetic Associations
MINING HIGH-LEVEL BRAIN IMAGING GENETIC ASSOCIATIONS Xiaohui Yao Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the School of Informatics and Computing, Indiana University March 2018 Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Huanmei Wu, PhD, Chair Doctoral Committee Li Shen, PhD January 16, 2018 Shiaofen Fang, PhD Jingwen Yan, PhD ii c 2018 Xiaohui Yao iii DEDICATION Dedicated to my dear family for all their love and support along the way iv ACKNOWLEDGEMENTS First and foremost I would like to express my deepest gratitude to my advisor, Dr. Li Shen, for guiding and supporting my research during the past five years. He has provided me with great research insights and exceptional enthusiasm as well as encouragement throughout my PhD study. This work would never be materialized without him. I also give my thanks to all the committee members for supporting this thesis work and all the constructive suggestions and feedback, that have been very helpful to keep this work on track and finally make the goal accomplished timely. I would like to express my appreciation to all my colleagues and professors from Center for Neuroimaging: Dr. Andrew J. Saykin, Dr. Shannon L. Risacher, Dr. Kwangsik Nho, and many others, who have provided me very valuable domain exper- tise from neurological, biological and genetic perspectives, as well as many invaluable medical data sources. I have learned considerably from their multi-perspective in- sights into problems. -
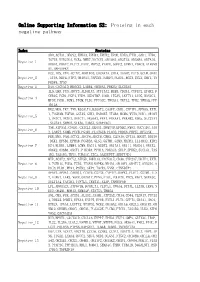
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Characterization of the Genomic Features and Expressed Fusion Genes In
1 SUPPLEMENTARY INFORMATION (ONLINE SUPPORTING INFORMATION) Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast Natrajan et al. Supplementary Methods Supplementary Figures S1-S6 Supplementary Tables S1-S7 2 SUPPLEMENTARY METHODS Tumor samples Two cohorts of micropapillary carcinomas (MPCs) were analyzed; the first cohort comprised 16 consecutive formalin fixed paraffin embedded (FFPE) MPCs, 11 pure and 5 mixed, which were retrieved from the authors' institutions (Table 1), and a second, validation cohort comprised 14 additional consecutive FFPE MPCs, retrieved from Molinette Hospital, Turin, Italy. Frozen samples were available from five out of the 16 cases from the first cohort of MPCs. As a comparator for the results of the Sequenom mutation profiling, a cohort of 16 consecutive IC-NSTs matched to the first cohort of 16 MPCs according to ER and HER2 status and histological grade were retrieved from a series of breast cancers previously analyzed by aCGH[1]. In addition, 14 IC-NSTs matched according to grade, and ER and HER2 status to tumors from the second cohort of 14 MPCs, and 48 grade 3 IC-NSTs were retrieved from Hospital La Paz, Madrid, Spain[1] (Supplementary Table S1). Power calculation For power calculations, we have assumed that if MPCs were driven by a recurrent fusion gene in a way akin to secretory carcinomas (which harbor the ETV6-NTRK3 fusion gene in >95% of cases[2-4]) or adenoid cystic carcinomas of the breast (which harbor the MYB-NFIB fusion gene in >90% of cases[5]), a ‘pathognomonic’ driver event would be present in at least ≥70% of cases (an estimate that is conservative). -

A General Statistic Framework for Genome-Based Disease Risk Prediction
A General Statistic Framework for Genome-based Disease Risk Prediction Long Ma1, Nan Lin1 and Momiao Xiong1,* 1 Division of Biostatistics, The University of Texas School of Public Health, Houston, TX 77030, USA Running Title: biomarker identification for risk prediction Key Words: genetic variants, clinical utility, sufficient dimension reduction, risk prediction, convex optimization. *Address for correspondence and reprints: Dr. Momiao Xiong, Human Genetics Center, The University of Texas Health Science Center at Houston, P.O. Box 20186, Houston, Texas 77225, (Phone): 713-500-9894, (Fax): 713-500-0900, E-mail: [email protected]. 1 Abstract Fast and more economical next generation sequencing (NGS) technologies will generate unprecedentedly massive and highly-dimensional genomic and epigenomic variation data. In the near future, a routine part of medical records will include the sequenced genomes. How to efficiently extract biomarkers for risk prediction and treatment selection from millions or dozens of millions of genomic variants raises a great challenge. Traditional paradigms for identifying variants of clinical validity are to test association of the variants. However, significantly associated genetic variants may or may not be usefulness for diagnosis and prognosis of diseases. Alternative to association studies for finding genetic variants of predictive utility is to systematically search variants that contain sufficient information for phenotype prediction. To achieve this, we introduce concepts of sufficient dimension reduction (SDR) and coordinate hypothesis which project the original high dimensional data to very low dimensional space while preserving all information on response phenotypes. We then formulate a clinically significant genetic variant discovery problem into the sparse SDR problem and develop algorithms that can select significant genetic variants from up to or even ten millions of predictors with the aid of a split-and-conquer approach. -

Research/0018.1
http://genomebiology.com/2001/2/6/research/0018.1 Research The human olfactory receptor repertoire comment Sergey Zozulya, ernando Echeverri and Trieu Nguyen Address: Senomyx Inc., 11099 North Torrey Pines Road, La Jolla, CA 92037, USA. Correspondence: Sergey Zozulya. E-mail: [email protected] reviews Published: 1 June 2001 Received: 8 March 2001 Revised: 12 April 2001 Genome Biology 2001, 2(6):research0018.1–0018.12 Accepted: 18 April 2001 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2001/2/6/research/0018 © 2001 Zozulya et al., licensee BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914) reports Abstract Background: The mammalian olfactory apparatus is able to recognize and distinguish thousands of structurally diverse volatile chemicals. This chemosensory function is mediated by a very large family of seven-transmembrane olfactory (odorant) receptors encoded by approximately 1,000 genes, the majority of which are believed to be pseudogenes in humans. deposited research Results: The strategy of our sequence database mining for full-length, functional candidate odorant receptor genes was based on the high overall sequence similarity and presence of a number of conserved sequence motifs in all known mammalian odorant receptors as well as the absence of introns in their coding sequences. We report here the identification and physical cloning of 347 putative human full-length odorant receptor genes. Comparative sequence analysis of the predicted gene products allowed us to identify and define a number of consensus sequence motifs and structural features of this vast family of receptors.