Petition for a Writ of Mandamus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ADVISORY BOARDS Each Issue of Herbaigram Is Peer Reviewed by Various Members of Our Advisory Boards Prior to Publication
ADVISORY BOARDS Each issue of HerbaiGram is peer reviewed by various members of our Advisory Boards prior to publication . American Botanical Council Herb Research Dennis V. C. Awang, Ph.D., F.C.I.C., MediPiont Natural Gail B. Mahadr, Ph.D., Research Assistant Professor, Products Consul~ng Services, Ottowa, Ontario, Conodo Deportment o Medical Chemistry &Pharmacognosy, College of Foundation Pharmacy, University of Illinois, Chicago, Illinois Manuel F. Balandrin, R.Ph., Ph.D., Research Scien~st, NPS Rob McCaleb, President Pharmaceuticals, Salt LakeCity , Utah Robin J. Maries, Ph.D., Associate Professor of Botany, Brandon University, Brandon, Manitoba, Conodo Mi(hael J. Balidt, Ph.D., Director of the lns~tute of Econom ic Glenn Appelt, Ph.D., R.Ph., Author and Profess or Botany, the New York Botanical Gorden, Bronx, New York Dennis J. M(Kenna, Ph.D., Consulting Ethnophormocologist, Emeritus, University of Colorado, and with Boulder Beach Joseph M. Betz, Ph.D., Research Chemist, Center for Food Minneapolis, Minnesota Consulting Group Safety and Applied Nutri~on, Division of Natural Products, Food Daniel E. Moerman, Ph.D., William E. Stirton Professor of John A. Beutler, Ph.D., Natural Products Chemist, and Drug Administro~on , Washington, D.C. Anthropology, University of Michigon/ Deorbom, Dearborn, Michigan Notional Cancer Institute Donald J. Brown, N.D., Director, Natural Products Research Consultants; Faculty, Bastyr University, Seattle, Washington Samuel W. Page, Ph.D., Director, Division of Natural Products, Robert A. Bye, Jr., Ph.D., Professor of Ethnobotony, Notional University of Mexico Thomas J. Carlson, M.S., M.D., Senior Director, Center for Food Safety and Applied Nutri~on , Food and Drug Administro~on , Washington, D.C. -
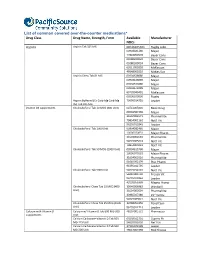
List of Common Covered Over-The-Counter Medications*
List of common covered over-the-counter medications* Drug Class Drug Name, Strength, Form Available Manufacturer NDCs Aspirin Aspirin Tab 325 MG 00536105301 Rugby Labo 00904681180 Major 12843054578 Bayer Cons 00280200020 Bayer Cons 00280200024 Bayer Cons 62011002003 McKesson 49348000110 McKes Sun Aspirin Chew Tab 81 MG 00904628880 Major 00904628889 Major 00904679480 Major 00904679489 Major 63739043401 McKesson 00536100836 Rugby Aspirin Buffered (Ca Carb-Mg Carb-Mg 70000014701 Leader Ox) Tab 325 MG Vitamin D3 supplements Cholecalciferol Tab 10 MCG (400 Unit) 00761005820 Basic Drug 00904582360 Major 31604002671 PharmaVite 79854001162 Nat’l Vit 96295012845 Leader Cholecalciferol Tab 1000 Unit 00904582460 Major 10006070033 Major Pharm 31604002683 PharmaVite 54629005024 Nat’l Vit 79854005023 Nat’l Vit Cholecalciferol Tab 50 MCG (2000 Unit) 00904615760 Major 10006070162 Major Pharm 31604002516 PharmaVite 51645092199 Plus Pharm 96295012795 Leader Cholecalciferol Tab 5000 Unit 54629794101 Nat’l Vit 58487003702 Freeda Vit 96295012846 Leader 43292056338 Magno-Hump Cholecalciferol Chew Tab 10 MCG (400 35046006982 Windwill Unit) 31604002604 PharmaVite 40985027380 21st Centu 54629089821 Nat’l Vit Cholecalciferol Chew Tab 25 MCG (1000 30768050356 Rexall Sun Unit) 96295012713 Leader Calcium with Vitamin D Calcium w/ Vitamin D Tab 600 MG-200 48107005122 Pharmassur supplements Unit Calcium Carbonate-Vitamin D Tab 500 60258012701 Cypress Ph MG-125 Unit 54629031630 Nat’l Vit Calcium Carbonate-Vitamin D Tab 500 37205043198 Leader MG-200 Unit 49614067298 -

Natural Medications for Psychiatric Disorders
Natural Medications for Psychiatric Disorders David Mischoulon, MD, PhD Director Depression Clinical and Research Program Massachusetts General Hospital Associate Professor of Psychiatry Harvard Medical School www.mghcme.org Disclosures • My spouse/partner and I have the following relevant financial relationship with a commercial interest to disclose: –Research support from Nordic Naturals –Unpaid consulting for Pharmavite, LLC, and Gnosis, Inc. –Royalties from Lippincott Williams & Wilkins for published book “Natural Medications for Psychiatric Disorders: Considering the Alternatives” www.mghcme.org 2 Objectives • To understand the evidence base for efficacy of natural therapies in psychiatry • To identify the risks and benefits of various natural treatments in psychiatry • To be able to educate patients in purchasing natural products in both over- the-counter and prescription forms www.mghcme.org Pros and Cons of Natural Remedies • In 2007, 38% of adults and 12% of children used CAM practices and products in the past year (NIH, 2010) – about $33.9 billion out-of-pocket cost • Easy access, good tolerability • Used by many who don’t respond to standard therapies • Limited research/systematic studies • “Natural” does NOT mean “safe” • Toxicity, adverse effects, interactions • Different preparations/purity • Insurance does not cover them www.mghcme.org 5 St. John’s Wort (SJW, Hypericum Perforatum) www.mghcme.org 6 St John’s Wort • About 40 published trials; many comparisons with TCAs and SSRIs; various systematic reviews and meta-analyses -

2016 What’S Hot 2016
OCTOBER 4-8 R Expo Hall October 6 & 7 2016 WHAT’S HOT 2016 THE WORLD'S LEADING INGREDIENT AND SOLUTIONS TRADESHOW WHERE SCIENCE & STRATEGY INTERSECT. WHAT’S HOT at SupplySide West TABLE OF CONTENTS A SPECIAL ALL-DIGITAL ISSUE October 2016 3 VIEWPOINT 4 AGENDA-AT-A-GLANCE 8 WHAT’S NEW IN CONTENT 9 SPORTS NUTRITION 10 SUPPLEMENTS & BEAUTY TABLE OF CONTENTS TABLE 11 LEGAL 12 FOOD & BEVERAGE 13 TARGETED CONTENT OPPORTUNITIES 16 SUPPLYSIDE CENTRAL 17 EDITOR’S CHOICE AWARDS 19 SURVIVAL KIT 20 FIRST-TIME ATTENDEE RECEPTION 22 EXHIBITOR NEWS 31 SUPPLYSIDE & VITAFOODS GLOBAL STOREFRONTS 32 HOT PRODUCTS Copyright © 2016 Informa Exhibitions LLC. All rights reserved. The publisher reserves the right to accept or reject any advertising or editorial material. Advertisers, and/or their agents, assume the responsibility for all content of published advertisements and assume responsibility for any claims against the publisher based on the advertisement. Editorial contributors assume responsibility for their published works and assume responsibility for any claims against the publisher based on the published work. Editorial content may not necessarily reflect the views of the publisher. Materials contained on this site may not be reproduced, modified, distributed, republished or hosted (either directly or by linking) without our prior written permission. You may not alter or remove any trademark, copyright or other notice from copies of content. You may, however, download material from the site (one machine readable copy and one print copy per page) for your personal, noncommercial use only. We reserve all rights in and title to all material downloaded. All items submitted to SUPPLYSIDE become the sole property of Informa Exhibitions LLC. -

Dietary Supplements Defined Dietary Supplement Research Facts And
Introduction to Dietary Supplements Since 1994, many non-traditional food components have been launched under the Dietary Supplements Health Education Act (DSHEA). Some of these include fish oil (a source of omega-3 fatty acids), glucosamine and chondroitin for joint health, and s-adenosylmethionine (SAMe) for depression, osteoarthritis and some liver diseases. Dietary Supplements Defined The DSHEA determined that dietary supplements would be defined by the following criteria: 1. A dietary supplement is "any product (except tobacco) that contains at least one of the following: (1) a vitamin, (2) a mineral, (3) an herb or botanical, (4) an amino acid, (5) a dietary substance for use by man to supplement the diet by increasing the total daily intake, or a concentrate, metabolite, constituent, extract, or combinations of these ingredients." 2. A dietary supplement is "intended for ingestion in pill, capsule, tablet or liquid form." 3. A dietary supplement is "not represented for use as a conventional food or as the sole item of a meal or diet." 4. A dietary supplement is "labeled as a 'dietary supplement'." 5. A dietary supplement "includes products such as an approved new drug, certified antibiotic, or licensed biologic that was marketed as a dietary supplement food before approval, certification, or license (unless the Secretary of Health and Human Services waives this provision)." Dietary Supplement Research Research on dietary supplements is growing each year, providing healthcare professionals with evidence of both safety and efficacy. For example, the U.S. Health and Human Services Department reported that SAMe is as effective as standard therapy for depression and osteoarthritis (HHS Hand Report 2002). -

(12) United States Patent (10) Patent No.: US 7.827,042 B2 Jung Et Al
US007827042B2 (12) United States Patent (10) Patent No.: US 7.827,042 B2 Jung et al. (45) Date of Patent: Nov. 2, 2010 (54) METHODS AND SYSTEMS RELATED TO a continuation-in-part of application No. 1 1/515,357, TRANSMISSION OF NUTRACEUTICAL filed on Sep. 1, 2006, and a continuation-in-part of ASSOCATED INFORMATION application No. 11/518,540, filed on Sep. 8, 2006, and a continuation-in-part of application No. 1 1/523,766, (75) Inventors: Edward K. Y. Jung, Bellevue, WA (US); filed on Sep. 18, 2006, and a continuation-in-part of Royce A. Levien, Lexington, MA (US); application No. 1 1/523,809, filed on Sep. 18, 2006, Robert W. Lord, Seattle, WA (US); and a continuation-in-part of application No. 1 1/637, Mark A. Malamud, Seattle, WA (US); 638, filed on Dec. 11, 2006, and a continuation-in-part John D. Rinaldo, Jr., Bellevue, WA of application No. 1 1/637,616, filed on Dec. 11, 2006, (US); Clarence T. Tegreene, Bellevue, and a continuation-in-part of application No. 1 1/314, WA (US); Lowell L. Wood, Jr., 945, filed on Dec. 20, 2005, and a continuation-in-part Bellevue, WA (US) of application No. 1 1/291,482, filed on Nov.30, 2005. (73) Assignee: The Invention Science Fund I, Inc. (51) Int. Cl. Bellevue, WA (US) G06O40/00 (2006.01) (*)c Notice: Subject to any disclaimer,- the term of this (58)(52) U.S.Field Cl. of Classification............................................... search ... 705/2; 705/2705/3 patent is extended or adjusted under 35 See application file for complete search history. -

A Strategic Analysis for New Product Entry Into the Nutracueticals, Functional Foods, and Vitamins and Supplements Markets
A STRATEGIC ANALYSIS FOR NEW PRODUCT ENTRY INTO THE NUTRACUETICALS, FUNCTIONAL FOODS, AND VITAMINS AND SUPPLEMENTS MARKETS Katherine Ray Bachelors of Science, University of Victoria, 1992 PROJECT SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF Business Administration In the Faculty of Business Administration 0 Katherine Ray 2004 SIMON FRASER UNIVERSITY August 2004 All rights reserved. This work may not be reproduced in whole or in part, by photocopy or other means, without permission of the author. APPROVAL Name: Katherine Ray Degree: Master of Business Administration Title of Project: A Strategic Analysis for New Product Entry into the Nutraceuticals, Functional Foods, and Vitamins and Supplements Markets Examining Committee: Jill S%epherd Assistant Professor,--, MO,T MBA Elicia Maine Assistant Professor, MOT MBA Date Approved: PARTIAL COPYRIGHT LICENCE I hereby grant to Simon Fraser University the right to lend my thesis, project or extended essay (the title of which is shown below) to users of the Simon Fraser University Library, and to make partial or single copies only for such users or in response to a request from the library of any other university, or other educational institution, on its own behalf or for one of its users. I further agree that permission for multiple copying of this work for schoiarly purposes may be granted by me or the Dean of Graduate Studies. It is understood that copying or publication of this work for financial gain shall not be allowed without my written permission. Title of Project: A Strategic Analysis for New Product Entry into the Nutraceuticals, Functional Foods, and Vitamins and Supplements Markets Author: Katherine Ray ABSTRACT Forbes Medi-Tech operates a biopharmaceutical company which is involved in research and development for the nutraceuticals and pharma industries. -

Supreme Court of the United States
No. _________ ================================================================ In The Supreme Court of the United States --------------------------------- --------------------------------- PHARMAVITE LLC, Petitioner, v. NOAH BRADACH, on behalf of himself and all others similarly situated, Respondent. --------------------------------- --------------------------------- On Petition For A Writ Of Certiorari To The United States Court Of Appeals For The Ninth Circuit --------------------------------- --------------------------------- PETITION FOR WRIT OF CERTIORARI --------------------------------- --------------------------------- DANIEL J. CONNOLLY FAEGRE BAKER DANIELS LLP 2220 Wells Fargo Center 90 South Seventh Street Minneapolis, MN 55402-3901 (612) 940-9453 [email protected] Counsel of Record for Petitioner ================================================================ COCKLE LEGAL BRIEFS (800) 225-6964 WWW.COCKLELEGALBRIEFS.COM i QUESTIONS PRESENTED 1. Is it a violation of the Supremacy Clause, U.S. CONST. art. VI, cl. 2, for a federal court to certify a class that includes class members asserting state law claims that are expressly preempted by federal statute, where the question of preemption cannot be determined for the class as a whole? 2. Is it a violation of the Supremacy Clause, U.S. CONST. art. VI, cl. 2, for a federal court to certify a class that includes class members asserting state law false labeling claims that are expressly preempted by fed- eral statute, if the federal court does not require indi- vidual -

Prenatal & Postnatal
PRENATAL & POSTNATAL Nutritional Support for Mother and Baby WHY DO I NEED A PRENATAL OR POSTNATAL SUPPLEMENT? A prenatal and postnatal multivitamin/mineral supplement provides essential daily nutritional support for pregnancy and lactation.† According to nationally representative research data, from diet alone Americans on average are failing to meet daily recommended levels of key nutrients such as vitamins A, C, D, E and K, calcium, magnesium, potassium and zinc, to name a few.2 Therefore, supplementation with a prenatal or postnatal multivitamin/mineral supplement helps address nutrient gaps to support the health of the mother and her growing baby.† Pregnancy and lactation place higher calorie and nutrient demands on the body, since the mother must meet the nutritional needs of a growing baby without sacrificing her own nutrient requirements. Multivitamin/mineral supplements designed for pregnancy or breastfeeding contain important nutrients to support the needs of both the mother and the growing baby, often including but not limited to folic acid, iron, calcium, vitamin D, iodine and DHA.† WHEN SHOULD I BEGIN TAKING A PRENATAL OR POSTNATAL SUPPLEMENT? A prenatal multivitamin supplement should be taken by women of childbearing age who are trying to conceive and throughout their pregnancy. The rationale for commencing the supplementation regimen prior to official pregnancy determination is to ensure the mother is receiving key nutrients needed even before she may know she is pregnant. For example, healthful diets with adequate folate/folic acid may reduce a woman’s risk of having a child with a brain or spinal cord defect. However, the neural tube is already formed by day 28 of gestation, before many women know they are pregnant.3 After childbirth, new mothers should take a daily postnatal multivitamin supplement. -

Vitamins and Supplements
vitamins and supplements Over $ 100 in savings inside 12ST0153_Answers_Brochure_Vitamins_v6.indd 1 4/6/12 2:34 PM Initial Date Project Title 12ST0153_Answers_Brochure_Vitamins_v6 On Strategy list Copy Editor Type of Piece Brochure On Brand Creative Mgr. Bleed .25 Creative Mgr. R. Rinka Has Impact Sr. Creative Mgr. Trim 5 x 7 Designer T. Jones Has Clarity Creative Director Creative Review Creative 4 pt. Check Team Setup Colors 4/4 Copywriter Status Final Output date 4/6/12 Revision # 6 beauty beauty Beauty Answers about vitamins and What nutrients should I consider for bones & joints Bones & Joints supplements begin here. promoting the health of my hair, skin The best way to get the daily requirement of essential vitamins and nails? is to eat a balanced diet that contains a variety of foods from We often appreciate and distinguish beauty based on an outer cognitive Cognitive the food guide pyramid. In addition to eating well, vitamins appearance, but much more can be said about beauty when and supplements may help ensure your body gets all the keeping our inner health in mind. Vitamins, minerals and other nutrients it needs. Since these needs can vary depending on key nutrients play a very important role in maintaining healthy age, gender and other factors, such as pregnancy, we created hair, skin and nails — the outward representation of our digestive Digestive this helpful guide to vitamins and supplements. “beauty” within. Read on to learn how the Contents following nutrients can help heart nourish and maintain the Heart Beauty .....................................Page 3 health of your hair, Bones & joints .........................Page 8 skin and nails. -

Dietary Supplement Labeling: Cognitive Biases, Market Manipulation & Consumer Choice
University of New Hampshire University of New Hampshire Scholars' Repository University of New Hampshire – Franklin Pierce Law Faculty Scholarship School of Law 1-1-2014 Dietary Supplement Labeling: Cognitive Biases, Market Manipulation & Consumer Choice Michael McCann University of New Hampshire School of Law Follow this and additional works at: https://scholars.unh.edu/law_facpub Part of the Alternative and Complementary Medicine Commons, Consumer Protection Law Commons, Food and Drug Law Commons, Marketing Law Commons, Other Nutrition Commons, and the Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Michael McCann, "Dietary Supplement Labeling: Cognitive Biases, Market Manipulation & Consumer Choice," 31 B. U. AM. J. L. & MED. 215 (2005). This Article is brought to you for free and open access by the University of New Hampshire – Franklin Pierce School of Law at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Law Faculty Scholarship by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. American Journalof Law & Medicine, 31 (2005): 215-268 © 2005 American Society of Law, Medicine & Ethics Boston University School of Law Dietary Supplement Labeling: Cognitive Biases, Market Manipulation & Consumer Choice Michael A. McCannt I. INTRODUCTION There exists increasing concern that the Dietary Supplements Health and Education Act ("DSHEA") has proven ineffective and perhaps counterproductive. Most illustratively, consider a recent and remarkably candid remark from Tommy Thompson, then Secretary of Health and Human Services: "I really think Congress should take a look at the food supplement law again. It doesn't make any sense to me." A health law that makes no sense to the Secretary of Health of Health and Human Services should certainly draw the attention of academics and policy-makers alike. -

Vol. 20, Fall 2007
NUTRITIONAL SCIENCES NEWS DIVISION OF NUTRITIONAL SCIENCES • UNIVERSITY OF ILLINOIS AT URBANA-CHAMPAIGN Vol 20, Fall 2007 NOTE FROM THE DIRECTOR It is a pleasure to share with you the accomplishments of the Division of Nutritional Sciences, its faculty, students and alumni in the past year. It is an exciting time to be at the University of Illinois! In the past year, six new faculty members joined the Division, which has helped to enrich the depth and breadth of expertise in nutrition and its affi liated disciplines within the division. We recruited 12 exceptional new students from 8 undergraduate institutions, who are mentored by 10 different faculty members. We also have undertaken the development of “Disciplinary Concentrations” within the division to highlight the research strengths of the faculty and to provide a means to facilitate faculty and student interactions and cross-training opportunities. This year we bid adieu to Don Layman and Keith Singletary, who retired in summer 2007. Don and Keith have been long-time members of the division and, together, trained over 17 M.S. and 17 Ph.D. students and contributed to many of the core nutrition courses. They will be missed. The campus Strategic Plan emphasizes building capacity for translational research in health and wellness and the need to undertake interdisciplinary research and undergraduate and graduate training. The Division and the faculty in its affi liated departments are well positioned to take advantage of these emerging opportunities. The Division’s Strategic Plan aims to assist its faculty and students to capitalize upon these new funding and research initiatives.