Characterizing the Sphingomyelinase Pathway Triggered by PRIMA-1 Derivatives in Lung Cancer Cells with Differing P53 Status
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accumulation of P53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells
Research Article Accumulation of p53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells Subhra Mohapatra, Baoky Chu, Xiuhua Zhao, and W.J. Pledger Department of Interdisciplinary Oncology, H. Lee Moffitt Cancer Center and Research Institute and the University of South Florida Medical Center, Tampa, Florida Abstract activates caspase-9. Most drugs signal apoptosis through the Toward the goal of developing effective treatments for mitochondrial pathway. prostate cancers, we examined the effects of cyclin-dependent Proteins that modulate caspase activity and thus determine kinase inhibitors on the survival of prostate cancer cells. We whether cells live or die include the inhibitor of apoptosis proteins show that roscovitine, R-roscovitine, and CGP74514A (collec- (IAPs) and the Bcl-2 proteins. The IAP family includes cIAP-1, cIAP- tively referred to as CKIs) induce the apoptosis of LNCaP and 2, XIAP, and survivin (11). Of these proteins, XIAP is the most potent. LNCaP-Rf cells, both of which express wild-type p53. Apoptosis IAPs interact with and inhibit the activity of processed caspases; required caspase-9 and caspase-3 activity, and cytochrome c thus, they function as ‘‘brakes’’ that can impede the apoptotic accumulated in the cytosol of CKI-treated cells. Amounts of process once it begins. IAPs inactivate both initiator and effector caspases; caspase-9 and caspase-3 are IAP targets, whereas caspase- p53 increased substantially in CKI-treated cells, whereas amounts of the endogenous caspase inhibitor XIAP decreased. 8 is not (12). CKIs did not appreciably induce the apoptosis of LNCaP cells The Bcl-2 proteins are critical determinants of mitochondria- treated with pifithrin-A, which prevents p53 accumulation, or dependent caspase activation (13). -

Life, Death, and the Pursuit of Apoptosis
Downloaded from genesdev.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Life, death, and the pursuit of apoptosis Eileen White Center for Advanced Biotechnology and Medicine and Department of Biological Sciences and the Cancer Institute of New Jersey, Rutgers University, Piscataway, New Jersey 08854 USA Apoptosis or programmed cell death is a genetically con- (Oltvai et al. 1993). Bcl-2 is expressed widely during em- trolled response for cells to commit suicide. The symp- bryogenesis (LeBrun et al. 1993; Novack and Korsmeyer toms of apoptosis are viability loss accompanied by cy- 1994), and its expression becomes more restricted in toplasmic boiling, chromatin condensation, and DNA adult tissues to those requiring long-term survival (stem fragmentation (Wyllie 1980). Pathologists and develop- cells, postmitotic neurons, and proliferating zones; mental biologists have cataloged the occurrences of ap- Hockenbery et al. 1991). optosis for many years based on these defined morpho- Apoptosis plays a major role in normal development, logical features, but what has propelled apoptosis into and it was expected that gain- or loss-of-function of a the forefront of basic research has been the identification death inhibitor such as Bcl-2 would have a phenotype in of genes that control cell death and the appreciation of transgenic mice. Bcl-2 was expected to regulate apopto- the role of apoptosis in development and disease. Regu- sis in lymphoid cells because of the role of Bcl-2 in hu- lation of cell death is essential for normal development man B cell lymphoma. Targeted Bcl-2 overexpression to and is an important defense against viral infection and the lymphoid system extends normal B cell survival the emergence of cancer. -

Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives
cancers Review Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives Shirin Hafezi 1 and Mohamed Rahmani 1,2,* 1 Research Institute of Medical & Health Sciences, University of Sharjah, P.O. Box 27272 Sharjah, United Arab Emirates; [email protected] 2 Department of Basic Medical Sciences, College of Medicine, University of Sharjah, P.O. Box 27272 Sharjah, United Arab Emirates * Correspondence: [email protected]; Tel.: +971-6505-7759 Simple Summary: Apoptosis, a programmed form of cell death, represents the main mechanism by which cells die. Such phenomenon is highly regulated by the BCL-2 family of proteins, which includes both pro-apoptotic and pro-survival proteins. The decision whether cells live or die is tightly controlled by a balance between these two classes of proteins. Notably, the pro-survival Bcl-2 proteins are frequently overexpressed in cancer cells dysregulating this balance in favor of survival and also rendering cells more resistant to therapeutic interventions. In this review, we outlined the most important steps in the development of targeting the BCL-2 survival proteins, which laid the ground for the discovery and the development of the selective BCL-2 inhibitor venetoclax as a therapeutic drug in hematological malignancies. The limitations and future directions are also discussed. Abstract: The major form of cell death in normal as well as malignant cells is apoptosis, which is a programmed process highly regulated by the BCL-2 family of proteins. This includes the antiapoptotic proteins (BCL-2, BCL-XL, MCL-1, BCLW, and BFL-1) and the proapoptotic proteins, which can be divided into two groups: the effectors (BAX, BAK, and BOK) and the BH3-only proteins Citation: Hafezi, S.; Rahmani, M. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -

Expression of the Tumor Necrosis Factor Receptor-Associated Factors
Expression of the Tumor Necrosis Factor Receptor- Associated Factors (TRAFs) 1 and 2 is a Characteristic Feature of Hodgkin and Reed-Sternberg Cells Keith F. Izban, M.D., Melek Ergin, M.D, Robert L. Martinez, B.A., HT(ASCP), Serhan Alkan, M.D. Department of Pathology, Loyola University Medical Center, Maywood, Illinois the HD cell lines. Although KMH2 showed weak Tumor necrosis factor receptor–associated factors expression, the remaining HD cell lines also lacked (TRAFs) are a recently established group of proteins TRAF5 protein. These data demonstrate that consti- involved in the intracellular signal transduction of tutive expression of TRAF1 and TRAF2 is a charac- several members of the tumor necrosis factor recep- teristic feature of HRS cells from both patient and tor (TNFR) superfamily. Recently, specific members cell line specimens. Furthermore, with the excep- of the TRAF family have been implicated in promot- tion of TRAF1 expression, HRS cells from the three ing cell survival as well as activation of the tran- HD cell lines showed similar TRAF protein expres- scription factor NF- B. We investigated the consti- sion patterns. Overall, these findings demonstrate tutive expression of TRAF1 and TRAF2 in Hodgkin the expression of several TRAF proteins in HD. Sig- and Reed–Sternberg (HRS) cells from archived nificantly, the altered regulation of selective TRAF paraffin-embedded tissues obtained from 21 pa- proteins may reflect HRS cell response to stimula- tients diagnosed with classical Hodgkin’s disease tion from the microenvironment and potentially (HD). In a selective portion of cases, examination of contribute both to apoptosis resistance and cell HRS cells for Epstein-Barr virus (EBV)–encoded maintenance of HRS cells. -
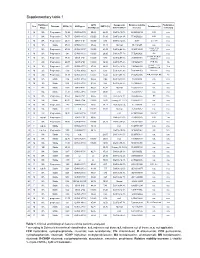
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Beyond Tumor Necrosis Factor Receptor: TRADD Signaling in Toll-Like Receptors
Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors Nien-Jung Chen*†‡, Iok In Christine Chio*‡, Wen-Jye Lin*, Gordon Duncan*, Hien Chau*§, David Katz*¶, Huey-Lan Huang*ʈ, Kelly A. Pike*,**, Zhenyue Hao*, Yu-Wen Su*, Kazuo Yamamoto*, Rene´ e F. de Pooter††, Juan Carlos Zu´ n˜ iga-Pflu¨ cker††, Andrew Wakeham*, Wen-Chen Yeh*, and Tak W. Mak*‡‡ *The Campbell Family Institute for Breast Cancer Research, Ontario Cancer Institute, University Health Network and Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 2C1; †Institute of Microbiology and Immunology, School of Life Science, National Yang-Ming University, Taipei, Taiwan 112, Republic of China; and ††Department of Immunology, University of Toronto, Sunnybrook and Women’s College Health Sciences Centre, 2075 Bayview Avenue, Toronto, ON, Canada M4N 3M5 Contributed by Tak W. Mak, July 9, 2008 (sent for review June 24, 2008) Tumor necrosis factor receptor 1-associated death domain protein tory responses in vivo, but also that this molecule is involved in (TRADD) is the core adaptor recruited to TNF receptor 1 (TNFR1) germinal center (GC) formation, T cell costimulation, and TLR upon TNF␣ stimulation. In cells from TRADD-deficient mice, TNF␣- signaling. Our TRADD knockout mice represent a very useful mediated apoptosis and TNF␣-stimulated NF-B, JNK, and ERK tool for extending the ever-increasing list of TRADD functions activation are defective. TRADD is also important for germinal in vitro and in vivo. center formation, DR3-mediated costimulation of T cells, and TNF␣- mediated inflammatory responses in vivo. TRADD deficiency does Results not enhance IFN␥-induced signaling. -

Dynamics of the BH3-Only Protein Binding Interface of Bcl-Xl
Biophysical Journal Volume 109 September 2015 1049–1057 1049 Article Dynamics of the BH3-Only Protein Binding Interface of Bcl-xL Xiaorong Liu,1 Alex Beugelsdijk,1 and Jianhan Chen1,* 1Department of Biochemistry and Molecular Biophysics, Kansas State University, Manhattan, Kansas ABSTRACT The balance and interplay between pro-death and pro-survival members of the B-cell lymphoma-2 (Bcl-2) family proteins play key roles in regulation of the mitochondrial pathway of programmed cell death. Recent NMR and biochemical studies have revealed that binding of the proapoptotic BH3-only protein PUMA induces significant unfolding of antiapoptotic Bcl-xL at the interface, which in turn disrupts the Bcl-xL/p53 interaction to activate apoptosis. However, the molecular mecha- nism of such regulated unfolding of Bcl-xL is not fully understood. Analysis of the existing Protein Data Bank structures of Bcl-xL in both bound and unbound states reveal substantial intrinsic heterogeneity at its BH3-only protein binding interface. Large-scale atomistic simulations were performed in explicit solvent for six representative structures to further investigate the intrinsic confor- mational dynamics of Bcl-xL. The results support that the BH3-only protein binding interface of Bcl-xL is much more dynamic compared to the rest of the protein, both unbound and when bound to various BH3-only proteins. Such intrinsic interfacial confor- mational dynamics likely provides a physical basis that allows Bcl-xL to respond sensitively to detailed biophysical properties of the ligand. The ability of Bcl-xL to retain or even enhance dynamics at the interface in bound states could further facilitate the regulation of its interactions with various BH3-only proteins such as through posttranslational modifications. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

The Molecular Links Between Cell Death and Inflammasome
cells Review The Molecular Links between Cell Death and Inflammasome Kwang-Ho Lee 1,2 and Tae-Bong Kang 1,2,* 1 Department of Biotechnology, College of Biomedical & Health Science, Konkuk University, Chungju 27478, Korea 2 Research Institute of Inflammatory Diseases, Konkuk University, Chungju 27478, Korea * Correspondence: [email protected]; Tel.: +82-43-840-3904; Fax: +82-43-852-3616 Received: 30 July 2019; Accepted: 9 September 2019; Published: 10 September 2019 Abstract: Programmed cell death pathways and inflammasome activation pathways can be genetically and functionally separated. Inflammasomes are specialized protein complexes that process pro-inflammatory cytokines, interleukin-1β (IL-1β), and IL-18 to bioactive forms for protection from a wide range of pathogens, as well as environmental and host-derived danger molecules. Programmed cell death has been extensively studied, and its role in the development, homeostasis, and control of infection and danger is widely appreciated. Apoptosis and the recently recognized necroptosis are the best-characterized forms of programmed death, and the interplay between them through death receptor signaling is also being studied. Moreover, growing evidence suggests that many of the signaling molecules known to regulate programmed cell death can also modulate inflammasome activation in a cell-intrinsic manner. Therefore, in this review, we will discuss the current knowledge concerning the role of the signaling molecules originally associated with programmed cell death in the activation of inflammasome and IL-1β processing. Keywords: inflammasome; apoptosis; necroptosis; programmed cell death; Caspase-8; RIPK1/3; MLKL; PGAM5; DRP1 1. Introduction Homeostasis is a principle property of living organisms and it is maintained at the systemic, tissue, and cellular levels through the homeostatic control system. -

SL10302R-FITC.Pdf
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-Hrk/FITC Conjugated antibody SL10302R-FITC Product Name: Anti-Hrk/FITC Chinese Name: FITC标记的BCL2相互作用蛋白质抗体 Activator of apoptosis harakiri; Activator of apoptosis Hrk; BCL2 interacting protein; BH3 interacting domain protein 3; BID3; DP5; Harakiri BCL2 interacting protein; Alias: Human brain 3UTR of mRNA for neuronal death protein partial sequence; Neuronal death protein DP5; HRK_HUMAN. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Cow, ICC=1:50-200IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 10kDa Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLH conjugated synthetic peptide derived from human Activator of apoptosis harakiri Lsotype: IgG Purification: affinitywww.sunlongbiotech.com purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: This gene encodes a member of the BCL-2 protein family. Members of this family are involved in activating or inhibiting apoptosis. The encoded protein localizes to Product Detail: intracellular membranes. This protein promotes apoptosis by interacting with the apoptotic inhibitors BCL-2 and BCL-X(L) via its BH3 domain. -

Differential Gene Expression in Brain and Liver Tissue of Wistar Rats After
Article Differential Gene Expression in Brain and Liver Tissue of Wistar Rats after Rapid Eye Movement Sleep Deprivation Atul Pandey 1,2,* , Ryan Oliver 2 and Santosh K Kar 1,3,* 1 School of Biotechnology, Jawaharlal Nehru University, New Delhi 110067, India 2 Department of Ecology, Evolution, and Behavior, The Alexander Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel; [email protected] 3 Nano Herb Research Laboratory, Kalinga Institute of Industrial Technology (KIIT) Technology Bio Incubator, Campus-11, KIIT Deemed to be University, Bhubaneswar, Odisha 751024, India * Correspondence: [email protected] (A.P.); santoshkariis@rediffmail.com (S.K.K.); Tel.: +972-547301848 (A.P.); +91-9937085111 (S.K.K.) Received: 10 September 2020; Accepted: 21 October 2020; Published: 23 October 2020 Abstract: Sleep is essential for the survival of most living beings. Numerous researchers have identified a series of genes that are thought to regulate “sleep-state” or the “deprived state”. As sleep has a significant effect on physiology, we believe that lack of total sleep, or particularly rapid eye movement (REM) sleep, for a prolonged period would have a profound impact on various body tissues. Therefore, using the microarray method, we sought to determine which genes and processes are affected in the brain and liver of rats following nine days of REM sleep deprivation. Our findings showed that REM sleep deprivation affected a total of 652 genes in the brain and 426 genes in the liver. Only 23 genes were affected commonly, 10 oppositely, and 13 similarly across brain and liver tissue.