Life, Death, and the Pursuit of Apoptosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accumulation of P53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells
Research Article Accumulation of p53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells Subhra Mohapatra, Baoky Chu, Xiuhua Zhao, and W.J. Pledger Department of Interdisciplinary Oncology, H. Lee Moffitt Cancer Center and Research Institute and the University of South Florida Medical Center, Tampa, Florida Abstract activates caspase-9. Most drugs signal apoptosis through the Toward the goal of developing effective treatments for mitochondrial pathway. prostate cancers, we examined the effects of cyclin-dependent Proteins that modulate caspase activity and thus determine kinase inhibitors on the survival of prostate cancer cells. We whether cells live or die include the inhibitor of apoptosis proteins show that roscovitine, R-roscovitine, and CGP74514A (collec- (IAPs) and the Bcl-2 proteins. The IAP family includes cIAP-1, cIAP- tively referred to as CKIs) induce the apoptosis of LNCaP and 2, XIAP, and survivin (11). Of these proteins, XIAP is the most potent. LNCaP-Rf cells, both of which express wild-type p53. Apoptosis IAPs interact with and inhibit the activity of processed caspases; required caspase-9 and caspase-3 activity, and cytochrome c thus, they function as ‘‘brakes’’ that can impede the apoptotic accumulated in the cytosol of CKI-treated cells. Amounts of process once it begins. IAPs inactivate both initiator and effector caspases; caspase-9 and caspase-3 are IAP targets, whereas caspase- p53 increased substantially in CKI-treated cells, whereas amounts of the endogenous caspase inhibitor XIAP decreased. 8 is not (12). CKIs did not appreciably induce the apoptosis of LNCaP cells The Bcl-2 proteins are critical determinants of mitochondria- treated with pifithrin-A, which prevents p53 accumulation, or dependent caspase activation (13). -

Expression of the Tumor Necrosis Factor Receptor-Associated Factors
Expression of the Tumor Necrosis Factor Receptor- Associated Factors (TRAFs) 1 and 2 is a Characteristic Feature of Hodgkin and Reed-Sternberg Cells Keith F. Izban, M.D., Melek Ergin, M.D, Robert L. Martinez, B.A., HT(ASCP), Serhan Alkan, M.D. Department of Pathology, Loyola University Medical Center, Maywood, Illinois the HD cell lines. Although KMH2 showed weak Tumor necrosis factor receptor–associated factors expression, the remaining HD cell lines also lacked (TRAFs) are a recently established group of proteins TRAF5 protein. These data demonstrate that consti- involved in the intracellular signal transduction of tutive expression of TRAF1 and TRAF2 is a charac- several members of the tumor necrosis factor recep- teristic feature of HRS cells from both patient and tor (TNFR) superfamily. Recently, specific members cell line specimens. Furthermore, with the excep- of the TRAF family have been implicated in promot- tion of TRAF1 expression, HRS cells from the three ing cell survival as well as activation of the tran- HD cell lines showed similar TRAF protein expres- scription factor NF- B. We investigated the consti- sion patterns. Overall, these findings demonstrate tutive expression of TRAF1 and TRAF2 in Hodgkin the expression of several TRAF proteins in HD. Sig- and Reed–Sternberg (HRS) cells from archived nificantly, the altered regulation of selective TRAF paraffin-embedded tissues obtained from 21 pa- proteins may reflect HRS cell response to stimula- tients diagnosed with classical Hodgkin’s disease tion from the microenvironment and potentially (HD). In a selective portion of cases, examination of contribute both to apoptosis resistance and cell HRS cells for Epstein-Barr virus (EBV)–encoded maintenance of HRS cells. -
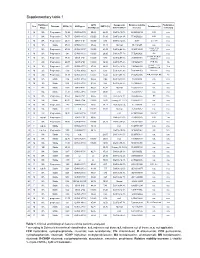
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Beyond Tumor Necrosis Factor Receptor: TRADD Signaling in Toll-Like Receptors
Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors Nien-Jung Chen*†‡, Iok In Christine Chio*‡, Wen-Jye Lin*, Gordon Duncan*, Hien Chau*§, David Katz*¶, Huey-Lan Huang*ʈ, Kelly A. Pike*,**, Zhenyue Hao*, Yu-Wen Su*, Kazuo Yamamoto*, Rene´ e F. de Pooter††, Juan Carlos Zu´ n˜ iga-Pflu¨ cker††, Andrew Wakeham*, Wen-Chen Yeh*, and Tak W. Mak*‡‡ *The Campbell Family Institute for Breast Cancer Research, Ontario Cancer Institute, University Health Network and Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 2C1; †Institute of Microbiology and Immunology, School of Life Science, National Yang-Ming University, Taipei, Taiwan 112, Republic of China; and ††Department of Immunology, University of Toronto, Sunnybrook and Women’s College Health Sciences Centre, 2075 Bayview Avenue, Toronto, ON, Canada M4N 3M5 Contributed by Tak W. Mak, July 9, 2008 (sent for review June 24, 2008) Tumor necrosis factor receptor 1-associated death domain protein tory responses in vivo, but also that this molecule is involved in (TRADD) is the core adaptor recruited to TNF receptor 1 (TNFR1) germinal center (GC) formation, T cell costimulation, and TLR upon TNF␣ stimulation. In cells from TRADD-deficient mice, TNF␣- signaling. Our TRADD knockout mice represent a very useful mediated apoptosis and TNF␣-stimulated NF-B, JNK, and ERK tool for extending the ever-increasing list of TRADD functions activation are defective. TRADD is also important for germinal in vitro and in vivo. center formation, DR3-mediated costimulation of T cells, and TNF␣- mediated inflammatory responses in vivo. TRADD deficiency does Results not enhance IFN␥-induced signaling. -

The Molecular Links Between Cell Death and Inflammasome
cells Review The Molecular Links between Cell Death and Inflammasome Kwang-Ho Lee 1,2 and Tae-Bong Kang 1,2,* 1 Department of Biotechnology, College of Biomedical & Health Science, Konkuk University, Chungju 27478, Korea 2 Research Institute of Inflammatory Diseases, Konkuk University, Chungju 27478, Korea * Correspondence: [email protected]; Tel.: +82-43-840-3904; Fax: +82-43-852-3616 Received: 30 July 2019; Accepted: 9 September 2019; Published: 10 September 2019 Abstract: Programmed cell death pathways and inflammasome activation pathways can be genetically and functionally separated. Inflammasomes are specialized protein complexes that process pro-inflammatory cytokines, interleukin-1β (IL-1β), and IL-18 to bioactive forms for protection from a wide range of pathogens, as well as environmental and host-derived danger molecules. Programmed cell death has been extensively studied, and its role in the development, homeostasis, and control of infection and danger is widely appreciated. Apoptosis and the recently recognized necroptosis are the best-characterized forms of programmed death, and the interplay between them through death receptor signaling is also being studied. Moreover, growing evidence suggests that many of the signaling molecules known to regulate programmed cell death can also modulate inflammasome activation in a cell-intrinsic manner. Therefore, in this review, we will discuss the current knowledge concerning the role of the signaling molecules originally associated with programmed cell death in the activation of inflammasome and IL-1β processing. Keywords: inflammasome; apoptosis; necroptosis; programmed cell death; Caspase-8; RIPK1/3; MLKL; PGAM5; DRP1 1. Introduction Homeostasis is a principle property of living organisms and it is maintained at the systemic, tissue, and cellular levels through the homeostatic control system. -

Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy
Review Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy Marilina García-Aranda 1, Elisabet Pérez-Ruiz 2 and Maximino Redondo 3,* 1 Research Unit, REDISSEC, Hospital Costa del Sol, Autovía A-7, km 187, 29603 Marbella, Málaga, Spain; [email protected] 2 Oncology Department, Hospital Costa del Sol, Autovía A-7, km 187, 29603 Marbella, Málaga, Spain; [email protected] 3 Research Unit, REDISSEC, Hospital Costa del Sol, Universidad de Málaga, Autovía A-7 km 187, 29603 Marbella, Málaga, Spain * Correspondence: [email protected] Received: 29 October 2018; Accepted: 6 December 2018; Published: 8 December 2018 Abstract: According to the World Health Organization (WHO), cancer is a leading cause of death worldwide. The identification of novel targets for cancer treatment is an area of intense work that has led Bcl-2 over-expression to be proposed as one of the hallmarks of cancer and Bcl-2 inhibition as a promising strategy for cancer treatment. In this review, we describe the different pathways related to programmed cell death, the role of Bcl-2 family members in apoptosis resistance to anti-cancer treatments, and the potential utility of Bcl-2 inhibitors to overcome resistance to chemo- and immunotherapy. Keywords: Bcl-2; resistance; apoptosis; inhibition; cancer; chemotherapy; immunotherapy 1. Introduction 1.1. Current Overview of Cancer Therapeutics Cancer is a multifactorial disease involving both genetic and environmental factors that has become one of the main health issues around the world. The figures, only for the year 2012, include 14.1 million of newly diagnosed cases and the death of 8.2 million people worldwide [1]. -

Caspase Activation & Apoptosis
RnDSy-lu-2945 Caspase Activation & Apoptosis Extrinsic & Intrinsic Pathways of Caspase Activation CASPASE CLEAVAGE & ACTIVATION Caspases are a family of aspartate-specific, cysteine proteases that serve as the primary mediators of Pro-Domain Large Subunit (p20) Small Subunit (p10) apoptosis. Mammalian caspases can be subdivided into three functional groups, apoptotic initiator TRAIL Pro-Domain α chain β chain caspases (Caspase-2, -8, -9, -10), apoptotic effector caspases (Caspase-3, -6, -7), and caspases involved Asp-x Asp-x Proteolytic cleavage in inflammatory cytokine processing (Caspase-1, -4, -5, 11, and -12L/12S). All caspases are synthesized α chain as inactive zymogens containing a variable length pro-domain, followed by a large (20 kDa) and a small Fas Ligand TRAIL R1 Pro-Domain β chain TRAIL R2 Heterotetramer Formation (10 kDa) subunit. FADD FADD α chain α chain Pro-caspase-8, -10 Active caspase Pro-caspase-8, -10 TWEAK β chain Apoptotic caspases are activated upon the receipt of either an extrinsic or an intrinsic death signal. The Fas/CD95 extrinsic pathway (green arrows) is initiated by ligand binding to cell surface death receptors (TNF RI, Fas/ MAMMALIAN CASPASE DOMAINS & CLEAVAGE SITES FADD FADD TNF-α APOPTOTIC CASPASES CD95, DR3, TRAIL R1/DR4, TRAIL R2/DR5) followed by receptor oligomerization and cleavage of Pro- Extrinsic Pathway DR3 (or another TWEAK R) INITIATOR CASPASES 152 316 331 Pro-caspase-8, -10 Pro-caspase-8, -10 1 435 caspase-8 and -10. Activation of Caspase-8 and Caspase-10 results in the cleavage of BID and Caspase-2 CARD TRADD FLIP TRADD downstream effector caspases. -

Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation
International Journal of Molecular Sciences Review Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation and Anti-Inflammation Jun-Hyuk Han 1, Jooho Park 1,2, Tae-Bong Kang 1,3,* and Kwang-Ho Lee 1,3 1 Department of Applied Life Sciences, Graduate School, BK21 Program, Konkuk University, Chungju 27478, Korea; [email protected] (J.-H.H.); [email protected] (J.P.); [email protected] (K.-H.L.) 2 Department of Biomedical Chemistry, College of Biomedical & Health Science, Konkuk University, Chungju 27487, Korea 3 Department of Biotechnology, College of Biomedical & Health Science, Konkuk University, Chungju 27487, Korea * Correspondence: [email protected]; Tel.: +82-43-840-3904 Abstract: Caspase-8 has been classified as an apoptotic caspase, and its initial definition was an initiator of extrinsic cell death. During the past decade, the concept of caspase-8 functioning has been changed by findings of its additional roles in diverse biological processes. Although caspase-8 was not originally thought to be involved in the inflammation process, many recent works have determined that caspase-8 plays an important role in the regulatory functions of inflammatory processes. In this review, we describe the recent advances in knowledge regarding the manner in which caspase-8 modulates the inflammatory responses concerning inflammasome activation, cell death, and cytokine induction. Keywords: caspase-8; inflammasome; inflammation; necroptosis; pyroptosis; apoptosis Citation: Han, J.-H.; Park, J.; Kang, T.-B.; Lee, K.-H. Regulation of Caspase-8 Activity at the Crossroads 1. Introduction of Pro-Inflammation and Anti-Inflammation. Int. J. Mol. Sci. Mammalian caspases have classically been divided into inflammatory and apoptotic 2021, 22, 3318. -

Death Signaling Paper Discussion • Apaf-1, a Human Protein
Hao Wu Death signaling References Paper Discussion • 1. Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev 15, 2922-2933. (2001). • 2. Fesik, S. W. Insights into programmed cell death through structural biology. Cell 103, 273- • Apaf-1, a human protein homologous to C. elegans CED-4, 282. (2000). participates in cytochrome c-dependent activation of • 3. Goyal, L. Cell death inhibition: keeping caspases in check. Cell 104, 805-808. (2001). • 4. Green, D. R. Apoptotic pathways: the roads to ruin. Cell 94, 695-698. (1998). caspase-3. Cell 90: 405-13, 1997. • 5. Green, D. R. Apoptotic pathways: paper wraps stone blunts scissors. Cell 102, 1-4. (2000). • Cytochrome c and dATP-dependent formation of Apaf- • 6. Hengartner, M. O. Apoptosis: corralling the corpses. Cell 104, 325-328. (2001). 1/Caspase-9 complex initiates an apoptotic protease • 7. Huang, D. C. & Strasser, A. BH3-Only proteins-essential initiators of apoptotic cell death. cascade. Cell 91: 479-89, 1997. Cell 103, 839-842. (2000). • 8. Johnstone, R. W., Ruefli, A. A. & Lowe, S. W. Apoptosis: a link between cancer genetics and chemotherapy. Cell 108, 153-164. (2002). • 9. Shi, Y. A structural view of mitochondria-mediated apoptosis. Nat Struct Biol 8, 394-401. (2001). • Color PDF file of handouts can be found at Wu lab web-page: http://venus.med.cornell.edu Apoptosis: an orderly process of cellular suicide Apoptosis plays important roles in many biological processes • Apoptosis refers to the shedding of leaves from trees in Greek. It was first observed by Carl Vogt in 1842. The word ‘apoptosis’ was • Physiological conditions introduced by Kerr, Wyllie and Currie in 1972 to describe the kind of – An intrinsic and integral component of physiology, just like cell death that is distinct from a necrotic cell death. -

Characterizing the Sphingomyelinase Pathway Triggered by PRIMA-1 Derivatives in Lung Cancer Cells with Differing P53 Status
ANTICANCER RESEARCH 34: 3271-3284 (2014) Characterizing the Sphingomyelinase Pathway Triggered by PRIMA-1 Derivatives in Lung Cancer Cells with Differing p53 Status EROICA SOANS1, SUSAN C. EVANS2, CYNTHIA CIPOLLA3* and ELROY FERNANDES4** 1,2,3,4Department of Chemistry and Biochemistry, Konneker Research Laboratories, Ohio University, The Ridges-Ohio University, Athens, OH, U.S.A. Abstract. Background/Aim: Derivatives of PRIMA-1 (hydroxymethyl)-1-azabicyclo [2,2,2] octan-3-one. It restores compound, 8a and 8b have been shown to increase mutant p53 activity in cell lines expressing His 175 mutant cytotoxicity in lung cancer cells through sphingomyelinase p53. This mutation causes major structural defect, as wild- pathways in IR and 8a or 8b co-treated lung cancer cells. type Arg 175 is required for stabilizing the DNA binding The goal of the present study was to further elaborate the domain structure and is frequently mutated and linked to molecular mechanism of 8a- or 8b-treated lung cancer cells improper protein folding. PRIMA-1 acts by forming adducts in order to understand their potential as anti-cancer drugs. with the thiols present on p53 through covalent modification, Materials and Methods: Biochemical assays, western blot, allowing for the correct folding of the p53 protein so that it flow cytometry and gene array analyses were employed to can bind DNA and transactivate downstream genes (1-4). distinguish these mechanisms. Results: Herein we PRIMA-1 is shown to increase the promoter activity and demonstrated that 8a and 8b cause apoptosis with S-phase protein expression of bax and PUMA, known transcriptional arrest in lung cancer cells by activating neutral targets of p53, and lower the expression of c-Jun and sphingomyelinase with ceramide production. -

A Novel Mechanism of TRAF Signaling Revealed by Structural and Functional Analyses of the TRADD–TRAF2 Interaction
Cell, Vol. 101, 777±787, June 23, 2000, Copyright 2000 by Cell Press A Novel Mechanism of TRAF Signaling Revealed by Structural and Functional Analyses of the TRADD±TRAF2 Interaction Young Chul Park,* Hong Ye,* Constance Hsia,² resistance, multiple organ failure, and neoplasm (Ash- Deena Segal,* Rebecca L. Rich,³ Hsiou-Chi Liou,² kenazi and Dixit, 1998; Leonen, 1998; Newton and De- David G. Myszka,³ and Hao Wu*§ cicco, 1999). *Department of Biochemistry The TNF receptor superfamily can be divided into ² Department of Medicine two subgroups, depending on whether the intracellular Weill Medical College and region contains a death domain. Receptors that contain Graduate School of Medical Sciences of death domains are known as death receptors (Nagata, Cornell University 1997; Ashkenazi and Dixit, 1998). The best studied death New York, New York 10021 receptors are TNFR1 and Fas; while TNFR1 only induces ³ Oncological Sciences cell death under certain circumstances and more often Huntsman Cancer Institute induces transcriptional gene activation, Fas is efficient University of Utah in cell death induction. TNF receptors that do not contain Salt Lake City, Utah 84132 death domains are represented by TNFR2, CD40, CD30, and many others. These receptors are involved primarily in gene transcription for cell survival, growth, and differ- entiation. Summary The cell activation, cell survival, and antiapoptotic functions of the TNF receptor superfamily are mostly TRAF proteins are major mediators for the cell activa- mediated by the family of TNF receptor±associated fac- tion, cell survival, and antiapoptotic functions of the tors (TRAF1±6) (Arch et al., 1998). The TRAF proteins TNF receptor superfamily. -

Apo2l/TRAIL-Dependent Recruitment of Endogenous FADD and Caspase-8 to Death Receptors 4 and 5
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Immunity, Vol. 12, 611±620, June, 2000, Copyright 2000 by Cell Press Apo2L/TRAIL-Dependent Recruitment of Endogenous FADD and Caspase-8 to Death Receptors 4 and 5 Frank C. Kischkel,*k David A. Lawrence,*k homophilic DD interactions. In turn, FADD recruits the Anan Chuntharapai,² Peter Schow,³ K. Jin Kim,² zymogen form of the apoptosis-initiating protease cas- and Avi Ashkenazi*§ pase-8, through homophilic interaction of ªdeath ef- *Department of Molecular Oncology fector domains.º The proximity of caspase-8 zymogens ² Department of Antibody Technology facilitates activation through self-processing, leading to ³ Department of Cell Biology cleavage of downstream effector caspases that execute Genentech, Inc. the apoptotic death program. 1 DNA Way The mechanism of apoptosis signaling by TNFR1 is South San Francisco, California 94080 similar to that of Fas, though not identical (Ashkenazi and Dixit, 1998). Upon TNF binding, TNFR1 recruits the adaptor TRADD, through homophilic DD interactions. Summary TRADD recruits FADD and caspase-8, initiating apo- ptosis. Alternatively, TRADD recruits the DD-containing Fas (APO-1/CD95) and tumor necrosis factor receptor adaptor RIP to activate NF-B and the adaptor TRAF-2 1 (TNFR1) trigger apoptosis by recruiting the apoptosis to activate the JNK/AP-1 pathway. Cells from trans- initiator caspase-8 through the adaptor FADD. Fas genic mice expressing dominant-negative FADD mutants binds FADD directly, whereas TNFR1 binds FADD indi- (FADD-DN) or from FADD gene knockout mice resist rectly, through TRADD.