The Molecular Links Between Cell Death and Inflammasome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accumulation of P53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells
Research Article Accumulation of p53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells Subhra Mohapatra, Baoky Chu, Xiuhua Zhao, and W.J. Pledger Department of Interdisciplinary Oncology, H. Lee Moffitt Cancer Center and Research Institute and the University of South Florida Medical Center, Tampa, Florida Abstract activates caspase-9. Most drugs signal apoptosis through the Toward the goal of developing effective treatments for mitochondrial pathway. prostate cancers, we examined the effects of cyclin-dependent Proteins that modulate caspase activity and thus determine kinase inhibitors on the survival of prostate cancer cells. We whether cells live or die include the inhibitor of apoptosis proteins show that roscovitine, R-roscovitine, and CGP74514A (collec- (IAPs) and the Bcl-2 proteins. The IAP family includes cIAP-1, cIAP- tively referred to as CKIs) induce the apoptosis of LNCaP and 2, XIAP, and survivin (11). Of these proteins, XIAP is the most potent. LNCaP-Rf cells, both of which express wild-type p53. Apoptosis IAPs interact with and inhibit the activity of processed caspases; required caspase-9 and caspase-3 activity, and cytochrome c thus, they function as ‘‘brakes’’ that can impede the apoptotic accumulated in the cytosol of CKI-treated cells. Amounts of process once it begins. IAPs inactivate both initiator and effector caspases; caspase-9 and caspase-3 are IAP targets, whereas caspase- p53 increased substantially in CKI-treated cells, whereas amounts of the endogenous caspase inhibitor XIAP decreased. 8 is not (12). CKIs did not appreciably induce the apoptosis of LNCaP cells The Bcl-2 proteins are critical determinants of mitochondria- treated with pifithrin-A, which prevents p53 accumulation, or dependent caspase activation (13). -

Inflammasome Activation-Induced Hypercoagulopathy
cells Review Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients Lealem Gedefaw, Sami Ullah, Polly H. M. Leung , Yin Cai, Shea-Ping Yip * and Chien-Ling Huang * Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Kowloon, Hong Kong, China; [email protected] (L.G.); [email protected] (S.U.); [email protected] (P.H.M.L.); [email protected] (Y.C.) * Correspondence: [email protected] (S.-P.Y.); [email protected] (C.-L.H.) Abstract: Coronavirus disease 2019 (COVID-19) is the most devastating infectious disease in the 21st century with more than 2 million lives lost in less than a year. The activation of inflammasome in the host infected by SARS-CoV-2 is highly related to cytokine storm and hypercoagulopathy, which significantly contribute to the poor prognosis of COVID-19 patients. Even though many studies have shown the host defense mechanism induced by inflammasome against various viral infections, mechanistic interactions leading to downstream cellular responses and pathogenesis in COVID-19 remain unclear. The SARS-CoV-2 infection has been associated with numerous cardiovascular disor- ders including acute myocardial injury, myocarditis, arrhythmias, and venous thromboembolism. The inflammatory response triggered by the activation of NLRP3 inflammasome under certain car- diovascular conditions resulted in hyperinflammation or the modulation of angiotensin-converting enzyme 2 signaling pathways. Perturbations of several target cells and tissues have been described in inflammasome activation, including pneumocytes, macrophages, endothelial cells, and dendritic cells. Citation: Gedefaw, L.; Ullah, S.; Leung, P.H.M.; Cai, Y.; Yip, S.-P.; The interplay between inflammasome activation and hypercoagulopathy in COVID-19 patients is an Huang, C.-L. -

Inflammasome Function in Neutrophils Kaiwen Chen Bachelor of Science (Honours Class I)
Inflammasome function in neutrophils Kaiwen Chen Bachelor of Science (Honours Class I) A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2015 Institute for Molecular Bioscience i ii Abstract The innate immune system protects against infection but also drives inflammatory disorders. Key molecular drivers of both processes are ‘inflammasomes’, multi-protein complexes that assemble in the cytosol to activate the protease, caspase-1. Active caspase-1 cleaves specific proinflammatory cytokines [e.g. interleukin (IL)-1β] into their mature, secreted forms, and initiates a form of inflammatory cell lysis called pyroptosis. Inflammasomes are assembled by select pattern recognition receptors such as NLRC4, NLRP3, AIM2, or via a non-canonical pathway involving caspase-11. Whilst inflammasomes functions have been intensely researched, the cell types mediating inflammasome signalling in distinct in vivo settings were unclear. Neutrophils are one of the first cells to arrive to a site of infection or injury, and thus have the opportunity to detect inflammasome-activating molecules in vivo, but their ability to signal by inflammasome pathways had not been closely examined. This thesis offers a detailed investigation of NLRC4, NLRP3 and caspase-11 inflammasome signalling in neutrophils during in vitro or in vivo challenge with whole microbe, purified microbial components, or adjuvant. This thesis demonstrates that acute Salmonella infection triggered NLRC4-dependent caspase-1 activation and IL-1β processing in neutrophils, and neutrophils were a major cellular compartment for IL-1β production during acute Salmonella challenge in vivo. Importantly, neutrophils did not undergo pyroptotic cell death upon NLRC4 activation, allowing these cells to sustain IL-1β production at a site of infection without compromising their crucial inflammasome-independent antimicrobial effector functions. -

Life, Death, and the Pursuit of Apoptosis
Downloaded from genesdev.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Life, death, and the pursuit of apoptosis Eileen White Center for Advanced Biotechnology and Medicine and Department of Biological Sciences and the Cancer Institute of New Jersey, Rutgers University, Piscataway, New Jersey 08854 USA Apoptosis or programmed cell death is a genetically con- (Oltvai et al. 1993). Bcl-2 is expressed widely during em- trolled response for cells to commit suicide. The symp- bryogenesis (LeBrun et al. 1993; Novack and Korsmeyer toms of apoptosis are viability loss accompanied by cy- 1994), and its expression becomes more restricted in toplasmic boiling, chromatin condensation, and DNA adult tissues to those requiring long-term survival (stem fragmentation (Wyllie 1980). Pathologists and develop- cells, postmitotic neurons, and proliferating zones; mental biologists have cataloged the occurrences of ap- Hockenbery et al. 1991). optosis for many years based on these defined morpho- Apoptosis plays a major role in normal development, logical features, but what has propelled apoptosis into and it was expected that gain- or loss-of-function of a the forefront of basic research has been the identification death inhibitor such as Bcl-2 would have a phenotype in of genes that control cell death and the appreciation of transgenic mice. Bcl-2 was expected to regulate apopto- the role of apoptosis in development and disease. Regu- sis in lymphoid cells because of the role of Bcl-2 in hu- lation of cell death is essential for normal development man B cell lymphoma. Targeted Bcl-2 overexpression to and is an important defense against viral infection and the lymphoid system extends normal B cell survival the emergence of cancer. -

Apoptotic Threshold Is Lowered by P53 Transactivation of Caspase-6
Apoptotic threshold is lowered by p53 transactivation of caspase-6 Timothy K. MacLachlan*† and Wafik S. El-Deiry*‡§¶ʈ** *Laboratory of Molecular Oncology and Cell Cycle Regulation, Howard Hughes Medical Institute, and Departments of ‡Medicine, §Genetics, ¶Pharmacology, and Cancer Center, University of Pennsylvania School of Medicine, Philadelphia, PA 19104 Communicated by Britton Chance, University of Pennsylvania School of Medicine, Philadelphia, PA, April 23, 2002 (received for review January 11, 2002) Little is known about how a cell’s apoptotic threshold is controlled Inhibition of the enzyme reduces the sensitivity conferred by after exposure to chemotherapy, although the p53 tumor suppres- overexpression of p53. These results identify a pathway by which sor has been implicated. We identified executioner caspase-6 as a p53 is able to accelerate the apoptosis cascade by loading the cell transcriptional target of p53. The mechanism involves DNA binding with cell death proteases so that when an apoptotic signal is by p53 to the third intron of the caspase-6 gene and transactiva- received, programmed cell death occurs rapidly. tion. A p53-dependent increase in procaspase-6 protein level al- lows for an increase in caspase-6 activity and caspase-6-specific Materials and Methods Lamin A cleavage in response to Adriamycin exposure. Specific Western Blotting and Antibodies. Immunoblotting was carried out by inhibition of caspase-6 blocks cell death in a manner that correlates using mouse anti-human p53 monoclonal (PAb1801; Oncogene), with caspase-6 mRNA induction by p53 and enhances long-term rabbit anti-human caspase-3 (Cell Signaling, Beverly, MA), mouse survival in response to a p53-mediated apoptotic signal. -

NLRP3-Inflammasome Inhibition During Respiratory Virus Infection
viruses Article NLRP3-Inflammasome Inhibition during Respiratory Virus Infection Abrogates Lung Immunopathology and Long-Term Airway Disease Development Carrie-Anne Malinczak 1 , Charles F. Schuler 2,3, Angela J. Duran 1, Andrew J. Rasky 1, Mohamed M. Mire 1, Gabriel Núñez 1, Nicholas W. Lukacs 1,3 and Wendy Fonseca 1,* 1 Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA; [email protected] (C.-A.M.); [email protected] (A.J.D.); [email protected] (A.J.R.); [email protected] (M.M.M.); [email protected] (G.N.); [email protected] (N.W.L.) 2 Department of Internal Medicine, Division of Allergy and Clinical Immunology, University of Michigan, Ann Arbor, MI 48109, USA; [email protected] 3 Mary H. Weiser Food Allergy Center, University of Michigan, Ann Arbor, MI 48109, USA * Correspondence: [email protected] Abstract: Respiratory syncytial virus (RSV) infects most infants by two years of age. It can cause severe disease leading to an increased risk of developing asthma later in life. Previously, our group has shown that RSV infection in mice and infants promotes IL-1β production. Here, we characterized the role of NLRP3-Inflammasome activation during RSV infection in adult mice and neonates. We observed that the inhibition of NLRP3 activation using the small molecule inhibitor, MCC950, or in Citation: Malinczak, C.-A.; Schuler, genetically modified NLRP3 knockout (Nlrp3−/−) mice during in vivo RSV infection led to decreased C.F.; Duran, A.J.; Rasky, A.J.; Mire, lung immunopathology along with a reduced expression of the mucus-associated genes and reduced M.M.; Núñez, G.; Lukacs, N.W.; production of innate cytokines (IL-1β, IL-33 and CCL2) linked to severe RSV disease while leading to Fonseca, W. -

Human RIPK1 Deficiency Causes Combined Immunodeficiency and Inflammatory Bowel Diseases
Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases Yue Lia,1, Marita Führerb,1, Ehsan Bahramia,1, Piotr Sochac, Maja Klaudel-Dreszlerc, Amira Bouzidia, Yanshan Liua, Anna S. Lehlea, Thomas Magga, Sebastian Hollizecka, Meino Rohlfsa, Raffaele Concaa, Michael Fieldd, Neil Warnere,f, Slae Mordechaig, Eyal Shteyerh, Dan Turnerh,i, Rachida Boukarij, Reda Belbouabj, Christoph Walzk, Moritz M. Gaidtl,m, Veit Hornungl,m, Bernd Baumannn, Ulrich Pannickeb, Eman Al Idrissio, Hamza Ali Alghamdio, Fernando E. Sepulvedap,q, Marine Gilp,q, Geneviève de Saint Basilep,q,r, Manfred Hönigs, Sibylle Koletzkoa,i, Aleixo M. Muisee,f,i,t,u, Scott B. Snapperd,i,v,w, Klaus Schwarzb,x,2, Christoph Kleina,i,2, and Daniel Kotlarza,i,2,3 aDr. von Hauner Children’s Hospital, Department of Pediatrics, University Hospital, Ludwig-Maximilians-Universität (LMU) Munich, 80337 Munich, Germany; bThe Institute for Transfusion Medicine, University of Ulm, 89081 Ulm, Germany; cDepartment of Gastroenterology, Hepatology, Nutritional Disorders and Pediatrics, The Children’s Memorial Health Institute, 04730 Warsaw, Poland; dDivision of Gastroenterology, Hepatology and Nutrition, Boston Children’s Hospital, Boston, MA 02115; eSickKids Inflammatory Bowel Disease Center, Research Institute, Hospital for Sick Children, Toronto, ON M5G1X8, Canada; fCell Biology Program, Research Institute, Hospital for Sick Children, Toronto, ON M5G1X8, Canada; gPediatric Gastroenterology, Hadassah University Hospital, Jerusalem 91120, Israel; hThe Juliet Keidan -

Expression of the Tumor Necrosis Factor Receptor-Associated Factors
Expression of the Tumor Necrosis Factor Receptor- Associated Factors (TRAFs) 1 and 2 is a Characteristic Feature of Hodgkin and Reed-Sternberg Cells Keith F. Izban, M.D., Melek Ergin, M.D, Robert L. Martinez, B.A., HT(ASCP), Serhan Alkan, M.D. Department of Pathology, Loyola University Medical Center, Maywood, Illinois the HD cell lines. Although KMH2 showed weak Tumor necrosis factor receptor–associated factors expression, the remaining HD cell lines also lacked (TRAFs) are a recently established group of proteins TRAF5 protein. These data demonstrate that consti- involved in the intracellular signal transduction of tutive expression of TRAF1 and TRAF2 is a charac- several members of the tumor necrosis factor recep- teristic feature of HRS cells from both patient and tor (TNFR) superfamily. Recently, specific members cell line specimens. Furthermore, with the excep- of the TRAF family have been implicated in promot- tion of TRAF1 expression, HRS cells from the three ing cell survival as well as activation of the tran- HD cell lines showed similar TRAF protein expres- scription factor NF- B. We investigated the consti- sion patterns. Overall, these findings demonstrate tutive expression of TRAF1 and TRAF2 in Hodgkin the expression of several TRAF proteins in HD. Sig- and Reed–Sternberg (HRS) cells from archived nificantly, the altered regulation of selective TRAF paraffin-embedded tissues obtained from 21 pa- proteins may reflect HRS cell response to stimula- tients diagnosed with classical Hodgkin’s disease tion from the microenvironment and potentially (HD). In a selective portion of cases, examination of contribute both to apoptosis resistance and cell HRS cells for Epstein-Barr virus (EBV)–encoded maintenance of HRS cells. -
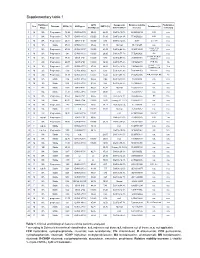
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

The Role of Caspase-2 in Regulating Cell Fate
cells Review The Role of Caspase-2 in Regulating Cell Fate Vasanthy Vigneswara and Zubair Ahmed * Neuroscience and Ophthalmology, Institute of Inflammation and Ageing, University of Birmingham, Birmingham B15 2TT, UK; [email protected] * Correspondence: [email protected] Received: 15 April 2020; Accepted: 12 May 2020; Published: 19 May 2020 Abstract: Caspase-2 is the most evolutionarily conserved member of the mammalian caspase family and has been implicated in both apoptotic and non-apoptotic signaling pathways, including tumor suppression, cell cycle regulation, and DNA repair. A myriad of signaling molecules is associated with the tight regulation of caspase-2 to mediate multiple cellular processes far beyond apoptotic cell death. This review provides a comprehensive overview of the literature pertaining to possible sophisticated molecular mechanisms underlying the multifaceted process of caspase-2 activation and to highlight its interplay between factors that promote or suppress apoptosis in a complicated regulatory network that determines the fate of a cell from its birth and throughout its life. Keywords: caspase-2; procaspase; apoptosis; splice variants; activation; intrinsic; extrinsic; neurons 1. Introduction Apoptosis, or programmed cell death (PCD), plays a pivotal role during embryonic development through to adulthood in multi-cellular organisms to eliminate excessive and potentially compromised cells under physiological conditions to maintain cellular homeostasis [1]. However, dysregulation of the apoptotic signaling pathway is implicated in a variety of pathological conditions. For example, excessive apoptosis can lead to neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, whilst insufficient apoptosis results in cancer and autoimmune disorders [2,3]. Apoptosis is mediated by two well-known classical signaling pathways, namely the extrinsic or death receptor-dependent pathway and the intrinsic or mitochondria-dependent pathway. -

NLRP1 Haplotypes Associated with Vitiligo and Autoimmunity Increase Interleukin-1Β Processing Via the NLRP1 Inflammasome
NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome Cecilia B. Levandowskia, Christina M. Maillouxa, Tracey M. Ferraraa, Katherine Gowana, Songtao Bena, Ying Jina,b, Kimberly K. McFannc, Paulene J. Hollanda, Pamela R. Faina,b,d, Charles A. Dinarelloe,1, and Richard A. Spritza,b,1 aHuman Medical Genetics and Genomics Program and bDepartment of Pediatrics, University of Colorado School of Medicine, Aurora, CO 80045; cColorado Biostatistics Consortium, Colorado School of Public Health, Aurora, CO 80045; and dBarbara Davis Center for Childhood Diabetes, and eDepartment of Medicine, University of Colorado School of Medicine, Aurora, CO 80045 Contributed by Charles A. Dinarello, January 8, 2013 (sent for review December 26, 2012) Nuclear localization leucine-rich-repeat protein 1 (NLRP1) is a key associated molecular patterns stimulate surface Toll-like recep- regulator of the innate immune system, particularly in the skin tors (TLRs) with subsequent assembly of the NLRP1 inflam- where, in response to molecular triggers such as pathogen- masome and activation of caspase-1. Active caspase-1 then associated or damage-associated molecular patterns, the NLRP1 cleaves the inactive IL-1β precursor (pro–IL-1β) to the mature inflammasome promotes caspase-1–dependent processing of bio- bioactive IL-1β (23), thereby stimulating downstream inflam- active interleukin-1β (IL-1β), resulting in IL-1β secretion and down- matory responses (23, 24). Because of its highly inflammatory stream inflammatory responses. NLRP1 is genetically associated and immunostimulating properties, the processing and release of with risk of several autoimmune diseases including generalized bioactive IL-1β are tightly controlled. However, in cryopyrin- vitiligo, Addison disease, type 1 diabetes, rheumatoid arthritis, associated periodic syndromes (CAPSs), which include familial and others.