Phosphinic Acids: Current Status and Potential for Drug Discovery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
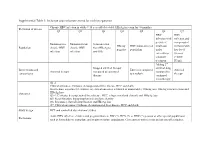
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Characterization of Atpase Activity of a Hepatitis C Virus NS3 Helicase Domain, and Analysis Involving Mercuric Reagents
J Biochem. 134, 505-511 (2003) DOI: 10.1093/jb/mvg 167 Characterization of ATPase Activity of a Hepatitis C Virus NS3 Helicase Domain, and Analysis Involving Mercuric Reagents Kiyoshi Kyono*,1, Masahiko Miyashiro2 and Ikuhiko Taguchi2 1 Medicinal Chemistry Research Laboratories and 2Discouery & Pharmacology Research Laboratories , Tanabe Seiyaku Co., Ltd., 16-89 Kashima 3-chome, Yodogawa-ku, Osaka 532-8505 Received March 31, 2003; accepted July 8, 2003 The C-terminal two-thirds of nonstructural protein 3 (NS3) of hepatitis C virus (HCV) exhibits RNA-dependent NTPase/helicase activity. This enzyme is considered to be involved in viral replication and is expected to be one of the target molecules of anti HCV drugs. In a search for NTPase inhibitors specific to HCV, we expressed and puri fied the truncated NS3 NTPase/helicase domain. Here, we report the characterization of its RNA-dependent ATPase activity. This enzyme preferred Mg2+ and the optimal pH was 7.0. We further investigated the effects of heavy metal ions on the ATPase activity. The mercuric ion inhibited it significantly, the 50% inhibitory concentration being 49 nM. The fact that the inhibitory profile was competitive and that this inhibi tion was blocked in the presence of a large excess of cysteine or dithiothreitol, sug gested that a cysteine residue in the DECH box was the main target site of mercury. Key words: ATPase activity, DEAD box protein, expression, HCV, NS3 helicase domain, mercuric reagent, purification. Abbreviations: BVDV, bovine viral diarrhea virus; YFV, yellow fever virus; CBB, Coomassie Brilliant Blue; MES, 2-(N-morpholino)ethanesulfonic acid; MOPS, morpholinepropanesulfonic acid; DTT, dithiothreitol; PCMB, ƒÏ-chlo romercuribenzoic acid. -

This Project Has Been Supported with Unrestriced Grants from Abbvie Gilead Sciences HEXAL Janssen-Cilag MSD Viiv Healthcare By
This project has been supported with unrestriced grants from AbbVie Gilead Sciences HEXAL Janssen-Cilag MSD ViiV Healthcare By Marcus Altfeld, Hamburg/Boston (USA) Achim Barmeyer, Dortmund Georg Behrens, Hannover Dirk Berzow, Hamburg Christoph Boesecke, Bonn Patrick Braun, Aachen Thomas Buhk, Hamburg Rob Camp, Barcelona (Spain/USA) Rika Draenert, Munich Christian Eggers, Linz (Austria) Stefan Esser, Essen Gerd Fätkenheuer, Cologne Gunar Günther, Windhoek (Namibia) Thomas Harrer, Erlangen Christian Herzmann, Borstel Christian Hoffmann, Hamburg Heinz-August Horst, Kiel Martin Hower, Dortmund Christoph Lange, Borstel Thore Lorenzen, Hamburg Tim Niehues, Krefeld Christian Noah, Hamburg Ramona Pauli, Munich Ansgar Rieke, Koblenz Jürgen Kurt Rockstroh, Bonn Thorsten Rosenkranz, Hamburg Bernhard Schaaf, Dortmund Ulrike Sonnenberg-Schwan, Munich Christoph D. Spinner, Munich Thomas Splettstoesser (Figures), Berlin Matthias Stoll, Hannover Hendrik Streeck, Essen/Boston (USA) Jan Thoden, Freiburg Markus Unnewehr, Dortmund Mechthild Vocks-Hauck, Berlin Jan-Christian Wasmuth, Bonn Michael Weigel, Schweinfurt Thomas Weitzel, Santiago (Chile) Eva Wolf, Munich HIV 2015/16 www.hivbook.com Edited by Christian Hoffmann and Jürgen K. Rockstroh Medizin Fokus Verlag IV Christian Hoffmann, M.D., Ph.D. ICH Stadtmitte (Infektionsmedizinisches Centrum Hamburg) Glockengiesserwall 1 20095 Hamburg, Germany Phone: + 49 40 2800 4200 Fax: + 49 40 2800 42020 [email protected] Jürgen K. Rockstroh, M.D., Ph.D. Department of Medicine I University of Bonn Sigmund-Freud-Strasse 25 53105 Bonn, Germany Phone: + 49 228 287 6558 Fax: + 49 228 287 5034 [email protected] HIV Medicine is an ever-changing field. The editors and authors of HIV 2015/16 have made every effort to provide information that is accurate and complete as of the date of publication. -

Synthetic Compounds with 2-Amino-1, 3, 4-Thiadiazole Moiety
molecules Review Synthetic Compounds with 2-Amino-1,3,4-Thiadiazole Moiety Against Viral Infections Georgeta Serban Pharmaceutical Chemistry Department, Faculty of Medicine and Pharmacy, University of Oradea, 29 Nicolae Jiga, 410028 Oradea, Romania; [email protected]; Tel.: +4-0756-276-377 Academic Editor: Antonio Carta Received: 29 January 2020; Accepted: 18 February 2020; Published: 19 February 2020 Abstract: Viral infections have resulted in millions of victims in human history. Although great efforts have been made to find effective medication, there are still no drugs that truly cure viral infections. There are currently approximately 90 drugs approved for the treatment of human viral infections. As resistance toward available antiviral drugs has become a global threat to health, there is an intrinsic need to identify new scaffolds that are useful in discovering innovative, less toxic and highly active antiviral agents. 1,3,4-Thiadiazole derivatives have been extensively studied due to their pharmacological profile, physicochemical and pharmacokinetic properties. This review provides an overview of the various synthetic compounds containing the 2-amino-1,3,4-thiadiazole moiety that has been evaluated for antiviral activity against several viral strains and could be considered possible prototypes for the development of new antiviral drugs. Keywords: 2-amino-1,3,4-thiadiazole; viral infections; antiviral agents; drug resistance; inhibitory concentration; mechanism of action 1. Human Viral Infections Viruses are the smallest among -

Genome Wide Mapping of Peptidases in Rhodnius Prolixus
ORIGINAL RESEARCH published: 12 December 2017 doi: 10.3389/fphys.2017.01051 Genome Wide Mapping of Peptidases in Rhodnius prolixus: Identification of Protease Gene Duplications, Horizontally Transferred Proteases and Analysis of Peptidase A1 Structures, with Considerations on Their Role in the Evolution of Edited by: Xanthe Vafopoulou, Hematophagy in Triatominae York University, Canada Reviewed by: Bianca S. Henriques 1†, Bruno Gomes 1†, Samara G. da Costa 1, Caroline da Silva Moraes 1, Leonardo Luis Fruttero, Rafael D. Mesquita 2, 3, Viv M. Dillon 4, Eloi de Souza Garcia 1, 2, Patricia Azambuja 1, 2, Facultad de Ciencias Químicas, 5 1, 2 Universidad Nacional de Córdoba, Roderick J. Dillon and Fernando A. Genta * Argentina 1 Laboratory of Insect Physiology and Biochemistry, Oswaldo Cruz Institute – Oswaldo Cruz Foundation (IOC-FIOCRUZ), Rio Márcio Galvão Pavan, de Janeiro, Brazil, 2 National Institute of Science and Technology for Molecular Entomology (INCT-EM), Cidade Universitária, Fundação Oswaldo Cruz (Fiocruz), Rio de Janeiro, Brazil, 3 Chemistry Institute, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil, 4 Institute of Integrative Brazil Biology, University of Liverpool, Liverpool, United Kingdom, 5 Division of Biomedical and Life Sciences, Lancaster University, *Correspondence: Lancaster, United Kingdom Fernando A. Genta genta@ioc.fiocruz.br; [email protected] Triatominae is a subfamily of the order Hemiptera whose species are able to feed in the †These authors have contributed vertebrate blood (i.e., hematophagy). This feeding behavior presents a great physiological equally to this work. challenge to insects, especially in Hemipteran species with a digestion performed by lysosomal-like cathepsins instead of the more common trypsin-like enzymes. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

(12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel Et Al
USOO8561811 B2 (12) United States Patent (10) Patent No.: US 8,561,811 B2 Bluchel et al. (45) Date of Patent: Oct. 22, 2013 (54) SUBSTRATE FOR IMMOBILIZING (56) References Cited FUNCTIONAL SUBSTANCES AND METHOD FOR PREPARING THE SAME U.S. PATENT DOCUMENTS 3,952,053 A 4, 1976 Brown, Jr. et al. (71) Applicants: Christian Gert Bluchel, Singapore 4.415,663 A 1 1/1983 Symon et al. (SG); Yanmei Wang, Singapore (SG) 4,576,928 A 3, 1986 Tani et al. 4.915,839 A 4, 1990 Marinaccio et al. (72) Inventors: Christian Gert Bluchel, Singapore 6,946,527 B2 9, 2005 Lemke et al. (SG); Yanmei Wang, Singapore (SG) FOREIGN PATENT DOCUMENTS (73) Assignee: Temasek Polytechnic, Singapore (SG) CN 101596422 A 12/2009 JP 2253813 A 10, 1990 (*) Notice: Subject to any disclaimer, the term of this JP 2258006 A 10, 1990 patent is extended or adjusted under 35 WO O2O2585 A2 1, 2002 U.S.C. 154(b) by 0 days. OTHER PUBLICATIONS (21) Appl. No.: 13/837,254 Inaternational Search Report for PCT/SG2011/000069 mailing date (22) Filed: Mar 15, 2013 of Apr. 12, 2011. Suen, Shing-Yi, et al. “Comparison of Ligand Density and Protein (65) Prior Publication Data Adsorption on Dye Affinity Membranes Using Difference Spacer Arms'. Separation Science and Technology, 35:1 (2000), pp. 69-87. US 2013/0210111A1 Aug. 15, 2013 Related U.S. Application Data Primary Examiner — Chester Barry (62) Division of application No. 13/580,055, filed as (74) Attorney, Agent, or Firm — Cantor Colburn LLP application No. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

A Abacavir Abacavirum Abakaviiri Abagovomab Abagovomabum
A abacavir abacavirum abakaviiri abagovomab abagovomabum abagovomabi abamectin abamectinum abamektiini abametapir abametapirum abametapiiri abanoquil abanoquilum abanokiili abaperidone abaperidonum abaperidoni abarelix abarelixum abareliksi abatacept abataceptum abatasepti abciximab abciximabum absiksimabi abecarnil abecarnilum abekarniili abediterol abediterolum abediteroli abetimus abetimusum abetimuusi abexinostat abexinostatum abeksinostaatti abicipar pegol abiciparum pegolum abisipaaripegoli abiraterone abirateronum abirateroni abitesartan abitesartanum abitesartaani ablukast ablukastum ablukasti abrilumab abrilumabum abrilumabi abrineurin abrineurinum abrineuriini abunidazol abunidazolum abunidatsoli acadesine acadesinum akadesiini acamprosate acamprosatum akamprosaatti acarbose acarbosum akarboosi acebrochol acebrocholum asebrokoli aceburic acid acidum aceburicum asebuurihappo acebutolol acebutololum asebutololi acecainide acecainidum asekainidi acecarbromal acecarbromalum asekarbromaali aceclidine aceclidinum aseklidiini aceclofenac aceclofenacum aseklofenaakki acedapsone acedapsonum asedapsoni acediasulfone sodium acediasulfonum natricum asediasulfoninatrium acefluranol acefluranolum asefluranoli acefurtiamine acefurtiaminum asefurtiamiini acefylline clofibrol acefyllinum clofibrolum asefylliiniklofibroli acefylline piperazine acefyllinum piperazinum asefylliinipiperatsiini aceglatone aceglatonum aseglatoni aceglutamide aceglutamidum aseglutamidi acemannan acemannanum asemannaani acemetacin acemetacinum asemetasiini aceneuramic -

Drug Interactions Between Hormonal Contraceptives and Antiretrovirals
Drug interactions between hormonal contraceptives and antiretrovirals Kavita Nandaa, Gretchen S. Stuartb, Jennifer Robinsonc, Andrew L. Grayd, Naomi K. Teppere and Mary E. Gaffieldf Objective: To summarize published evidence on drug interactions between hormonal contraceptives and antiretrovirals. Design: Systematic review of the published literature. Methods: We searched PubMed, POPLINE, and EMBASE for peer-reviewed publi- cations of studies (in any language) from inception to 21 September 2015. We included studies of women using hormonal contraceptives and antiretrovirals concurrently. Outcomes of interest were effectiveness of either therapy, toxicity, or pharmacokinetics. We used standard abstraction forms to summarize and assess strengths and weaknesses. Results: Fifty reports from 46 studies were included. Most antiretrovirals whether used for therapy or prevention, have limited interactions with hormonal contraceptive methods, with the exception of efavirenz. Although depot medroxyprogesterone acetate is not affected, limited data on implants and combined oral contraceptive pills suggest that efavirenz-containing combination antiretroviral therapy may compromise contraceptive effectiveness of these methods. However, implants remain very effective despite such drug interactions. Antiretroviral plasma concentrations and effectiveness are generally not affected by hormonal contraceptives. Conclusion: Women taking antiretrovirals, for treatment or prevention, should not be denied access to the full range of hormonal contraceptive options, but should be counseled on the expected rates of unplanned pregnancy associated with all contra- ceptive methods, in order to make their own informed choices. Copyright Q 2017 The Author(s). Published by Wolters Kluwer Health, Inc. AIDS 2017, 31:917–952 Keywords: antiretroviral therapy, contraceptive implant, depot medroxyprogesterone acetate, HIV, hormonal contraception, systematic review Introduction Decreasing unintended pregnancies also reduces vertical HIV transmission [5].