Hoffmann Rockstroh |
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Long-Acting Cabotegravir: the Future of HIV Prep
Long-Acting Injectable Cabotegravir: the Future of HIV PrEP? Brian R. Wood, MD Associate Professor of Medicine University of Washington Mountain West AIDS Education & Training Center June 4, 2020 Disclosures No conflicts of interest or relationships to disclose. Will be discussing an investigational antiretroviral. Full HPTN 083 study results not yet available. Will be reviewing data from a preliminary DSMB analysis today. See press release and webinar: https://www.hptn.org/news-and-events/announcements/cab- la-proves-be-highly-effective-prevention-hiv-acquisition Outline • General notes about cabotegravir • News from the phase 3 PrEP trial (and why it’s a big deal) • Questions, concerns, and next steps for long-acting PrEP What is Cabotegravir? Cabotegravir (CAB) • Investigational integrase strand transfer inhibitor • Potential infrequent dosing and parenteral administration - Oral half-life: 40 hours - Parenteral nanosuspension (IM, SC) half-life: 21-50 days - Median time from discontinuation to undetectable plasma level (IM, SC): 43-66 weeks • Metabolized by UGT1A1 (main pathway) & UGT1A9 - Minimal CYP metabolism; likely few drug interactions • Relatively low barrier to resistance Aidsinfo.nih.gov/drugs Injectable Long-Acting Cabotegravir Image courtesy of Dr. Raphael Landovitz, UCLA What is the HPTN 083 Trial and What’s the Big News? HPTN 083 A Phase 2b/3 Double Blind Safety and Efficacy Study of Injectable Cabotegravir Compared to Daily Oral TDF/FTC, for Pre-Exposure Prophylaxis in HIV-Uninfected Cisgender Men and TranHPTNsgender Women wh o 083have Sex wi thSites Men – Phase 2b/3 Target enrollment: 4,500 HIV- uninfected cisgender men and transgender 45 Sites in 8 Countrieswomen who have sex with men and who are at risk of HIV acquisition Primary outcome: HIV Prevention effectiveness of cabotegravir compared to daily oral TDF/FTC United States India Vietnam Thailand Peru Brazil South Argentina Africa ClinicalTrials.gov Identifier: NCT02720094 Slide courtesy of Dr. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0306098 A1 Mutz Et Al
US 200803 06098A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0306098 A1 Mutz et al. (43) Pub. Date: Dec. 11, 2008 (54) PHARMACOKINETICS OF PROTEASE Publication Classification INHIBITORS AND OTHER DRUGS (51) Int. Cl. A 6LX 3L/505 (2006.01) (76) Inventors: Mitchell W. Mutz, La Jolla, CA A63L/4353 (2006.01) (US); Jason E. Gestwicki, Ann C07D 49/12 (2006.01) Arbor, MI (US) C07D 239/04 (2006.01) A6IP3 L/18 (2006.01) Correspondence Address: (52) U.S. Cl. ............ 514/274: 514/291; 546/90; 54.4/316 MINTZ, LEVIN, COHN, FERRIS, GLOVSKY (57) ABSTRACT AND POPEO, PC 5 Palo Alto Square - 6th Floor,3000 El Camino Real A method for modulating at least one pharmacokinetic prop PALO ALTO, CA 94306-2155 (US) erty of a protease inhibitor upon administration to a host is provided. One administers to the host an effective amount of a bifunctional compound of less than about 5000 daltons (21) Appl. No.: 12/151,329 comprising the protease inhibitor or an active derivative thereof and a pharmacokinetic modulating moiety. The phar (22) Filed: May 5, 2008 macokinetic modulating moiety binds to at least one intrac ellular protein. The bifunctional compound has at least one (30) Foreign Application Priority Data modulated pharmacokinetic property upon administration to the hostas compared to a free drug control that comprises the Nov. 6, 2006 (US) ................. PCT/US2006/043400 protease inhibitor. FKBP-binding FKBP-binding interface interface Conjugate Target Binding Calcineurin-binding interface Patent Application Publication Dec. 11, 2008 Sheet 1 of 11 US 2008/0306.098 A1 O) \iO Ol cy A- S' O I N CD (5. -

HIV-Infected Patients
New Drugs for the Treatment-Experienced Patient Joseph Eron, md Associate Professor of Medicine and Director, Clinical Core unc Center for aids Research, University of North Carolina at Chapel Hill Summary by Tim Horn Edited by Jay Dobkin, md; Michael Saag, md reatment options for antiretro- humans by adenosine deaminase into D- Deeks and his colleagues in 1998 demon- viral-experienced patients leave a dioxolane guanine (dxg), a metabolite that strated a 1 log reduction in hiv-rna in hiv- lot to be desired. According to Dr. has potent activity against hiv and hbv. infected patients—more than 50% of whom Joe Eron, patients who have treat- According to in vitro data presented at were treatment-experienced—who received ment experience in all three classes the 3rd International Workshop on hiv tenofovir df 300 mg once daily as of currently available antiretrovi- Drug Resistance and Treatment Strategies, monotherapy for 28 days (Deeks, 1998). Trals have, at best, a 30% chance of re- held in June 1999, dapd was found to in- According to in vitro data presented by ducing their viral load to levels below 400 hibit wild-type and mutant isolates resistant Gilead’s Dr. Michael Miller at the recent copies/mL upon initiating a salvage regi- to azt (Retrovir) and 3TC (Borroto-Esoda, 4th International Resistance Workshop, the men. Cross-resistance within each class of 1999). The drug was also reported to be ac- resistance pattern for tenofovir df is simi- drugs, particularly the protease inhibitors tive against strains collected from patients lar to that of its chemical predecessor adefo- (pis) and non-nucleoside reverse tran- who have failed various nrti and nnrti vir, a compound no longer in development scriptase inhibitors (nnrtis), essentially combination therapies, including those for the treatment of hiv (Miller, 2000). -

LATTE Study Oral Cabotegravir + Rilpivirine Versus Efavirenz + 2 NRTI’S LATTE Study: Design
Oral Cabotegravir + Rilpivirine versus Efavirenz + 2 NRTI’s LATTE Study Oral Cabotegravir + Rilpivirine versus Efavirenz + 2 NRTI’s LATTE Study: Design Study Design: CAB 10 mg CAB 10 mg + 2 NRTI’s + RPV 25 mg • BacKground: Phase 2b, (n = 60) (n = 52) randoMized, partially blinded study done at Multiple centers CAB 30 mg CAB 30 mg in the U.S. and Canada + 2 NRTI’s + RPV 25 mg (n = 60) (n = 51) • Inclusion Criteria (n = 244) - Age ≥18 years - Antiretroviral-naïve CAB 60 mg CAB 60 mg - HIV RNA >1,000 copies/ML + 2 NRTI’s + RPV 25 mg - CD4 count >200 cells/MM3 (n = 61) (n = 53) - CrCl >50 ML/Min - No hepatitis B Efavirenz 600 mg Efavirenz 600 mg - No significant transaMinitis + 2 NRTI’s + 2 NRTI’s (n = 62) (n = 46) 24-week lead-in phase Source: Margolis DA, et al. Lancet Infect Dis. 2015;15:1145-55. Oral Cabotegravir + Rilpivirine versus Efavirenz + 2 NRTI’s LATTE Study: Results Cabotegravir + 2NRTIs Cabotegravir + Rilpivirine Efavirenz + 2NRTIs Induction* Maintenance* 100 80 86 82 76 74 71 60 63 40 HIV RNA <50 copies/mL (%) <50 copies/mL HIV RNA 20 156/181 46/62 149/181 44/62 137/181 39/62 0 Week 24 Week 48 Week 96 *Cabotegravir data is composite of all cabotegravir doses Source: Margolis DA, et al. Lancet Infect Dis. 2015;15:1145-55. Oral Cabotegravir + Rilpivirine versus Efavirenz + 2 NRTI’s LATTE Study: Results 100 Induction* Maintenance 80 60 Cabotegravir 10 mg + Rilpivirine 40 Cabotegravir 30 mg + Rilpivirine HIV RNA <40 copies/mL HIV RNA 20 Cabotegravir 60 mg + Rilpivirine Efavirenz 600 mg + 2NRTIs 0 0 12 24 36 48 60 72 84 96 Treatment Week *During induction phase cabotegravir administered with investigator chosen 2NRTIs Source: Margolis DA, et al. -
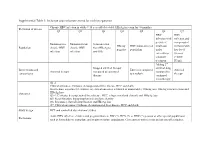
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

( 12 ) United States Patent
US010385067B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 , 385, 067 B2 Carra et al. (45 ) Date of Patent: Aug. 20 , 2019 (54 ) SODIUM (2R , 55 , 13AR ) - 7 , 9 -DIOX0 - 10 ( 56 ) References Cited ( 2 , 4 ,6 - TRIFLUOROBENZYL )CARBAMOYL ) 2 , 3 , 4 , 5 , 7 , 9 , 13 , 13A -OCTAHYDRO - 2 , 5 U . S . PATENT DOCUMENTS METHANOPYRIDO [ 1 ' , 2 ' : 4 , 5 ]PYRAZINO 5 , 814 ,639 A 9 / 1998 Liotta et al . [ 2 , 1 - B ] [ 1 , 3 ]OXAZEPIN - 8 - OLATE 5 , 914 , 331 A 6 / 1999 Liotta et al . 5 ,922 ,695 A 7 / 1999 Arimilli et al . 5 , 935 , 946 A 8 / 1999 Munger, Jr . et al. (71 ) Applicant: Gilead Sciences , Inc ., Foster Ctiy , CA 5 , 977 , 089 A 11/ 1999 Arimilli et al. (US ) 6 ,043 , 230 A 3 / 2000 Arimilli et al. 6 ,620 , 841 B1 9 / 2003 Fujishita et al . (72 ) Inventors : Ernest A . Carra , Foster City , CA ( US ) ; 6 ,642 , 245 B1 11/ 2003 Liotta et al. 6 , 703 , 396 B1 3 / 2004 Liotta et al . Irene Chen , San Mateo , CA (US ) ; 7 , 176 , 220 B2 2 /2007 Satoh et al. Vahid Zia , Palo Alto , CA (US ) 7 ,419 , 969 B2 9 / 2008 Naidu et al. 7 , 550 , 463 B2 6 / 2009 Yoshida (73 ) Assignee : Gilead Sciences , Inc. , Foster City , CA 7 ,635 , 704 B2 12 /2009 Satoh et al. 7 , 858 , 788 B2 12 / 2010 Yoshida et al . (US ) 8 , 129 , 385 B2 3 / 2012 Johns et al . 8 , 148 , 374 B2 4 / 2012 Desai et al. ( * ) Notice : Subject to any disclaimer , the term of this 8 , 188 , 271 B2 5 / 2012 Yoshida et al . -

Qualitative Study to Explore the Knowledge and Attitude of Pregnant Women Regarding HIV/AIDS Testing in Kotayk Region and in Yerevan, Armenia
Qualitative study to explore the knowledge and attitude of pregnant women regarding HIV/AIDS testing in Kotayk region and in Yerevan, Armenia Utilizing Professional Publication Framework Henrik Khachatryan, MD, MPH candidate, American University of Armenia Primary Adviser: M. Thompson, MS, DrPH Secondary Adviser: K. White, RN, PhD, CNAA October 2005 Table of Contents INTRODUCTION ...........................................................................................................................................................1 Background Information and Literature review....................................................................................................1 Rationale for the Research and Research Questions.............................................................................................4 METHODS AND MATERIALS........................................................................................................................................5 Study design...........................................................................................................................................................5 Study population ....................................................................................................................................................5 Sampling and Study Setting...................................................................................................................................6 Data collection Instrument.....................................................................................................................................6 -

The Design, Synthesis and Optimization of Allosteric Hiv-1
THE DESIGN, SYNTHESIS AND OPTIMIZATION OF ALLOSTERIC HIV-1 INTEGRASE INHIBITORS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Janet Antwi Graduate Program in Pharmaceutical Sciences The Ohio State University 2017 Dissertation Committee: Professor James R. Fuchs, Advisor Professor Werner Tjarks Professor Karl A. Werbovetz Copyright by Janet Antwi 2017 Abstract In the past quarter century, there has been tremendous progress in the discovery of antiretroviral therapy, making HIV/AIDS a manageable chronic disease. However, the HIV virus is relentless and continues to evolve under drug pressure to escape control and continue infection. The enzyme HIV integrase is responsible for the incorporation of viral double stranded DNA into a host chromosomal DNA and has recently become an attractive target in combating HIV resistance. Raltegravir (RAL), elvitegravir (EVG) and dolutegravir (DTG) are three clinically approved active-site integrase inhibitors. Unfortunately, mutations of the enzyme observed in patients have resulted in resistance to thedrug in the clinic. A new approach to targeting integrase (IN) is the development of allosteric inhibitors that specifically target the protein-protein interaction between IN and its cellular cofactor LEDGF/p75. Recently discovered quinoline-based allosteric integrase inhibitor (ALLINI) B1224436 was the first compound to advance into clinical trials but was discontinued due to poor pharmacokinetic properties including low in vivo clearance. In addition, several reports have revealed the emergence of resistance due to mutation to quinoline based ALLINIs. Applying scaffold hopping approach, several pyridine-based, thiophenes, ii pyrazoles, isoquinolines and other heteroaromatic cores have been studied as ALLINIs. -

Nnrtis – MK1439 New Classes • Maturation Inhibitors, LEDGINS, Etc
New Antiretrovirals and New Strategies Saye Khoo HIV Pharmacology Group Declaration of Interests •www.hiv-druginteractions.org & www.hep-druginteractions.org sponsorship from Janssen, ViiV, Abbott, Merck, BMS, Gilead, Boehringer, Vertex. Editorial content remains independent. •Research Grants: Merck, ViiV •Speakers bureau: Merck, Janssen, Abbott, Roche •Travel grants: Gilead, ViiV, BMS, Janssen •TaiLor trial (NIHR-funded) Antiretroviral Stewardship Myocardial Infarction Stroke Cancer Congnitive impairment Liver, renal etc Plan Now For Then • what to give ? • Minimise resistance • when to start ? • Minimise toxicity • how to manage ? • Preserve options • Normalise Immunity • Equip for future co-morbidity New Drugs, New Formulations, New Strategies Improvements on existing classes • TAF • dolutegravir and other integrases • new NNRTIs – MK1439 New Classes • Maturation inhibitors, LEDGINS, etc New Formulations • nanoformulations • mono- or dual therapy • LA injections or implants • targeting latent reservoirs New Strategies • NRTI-sparing, PI monotherapy • targeting latent reservoirs • targeting immune activation, cardiovascular risk • etc INSTIs NRTIs PIs NNRTIs Other Approved Dolutegravir Phase 3 TAF DRVc Doravirine TAF/FTC/EVGc (MK1349) Cenicriviroc RPV-LA BMS663068 Phase 2 GSK126744 Racivir ABC/3TC/DTG Amodoxovir TAF/FTC/DRVc Elvucitabine Doravirine (MK-1439) • Pharmacology – Potent - IC95 ~19 nM (50% human serum) – Once-daily dosing; T½ 10-16h – P450 metabolism (CYP3A4/5) • No significant inhibition/induction of CYP P450s • No significant -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

HEPP Report: Infectious Diseases in Corrections, Vol. 6 No. 6 HIV & Hepatitis Education Prison Project
University of Rhode Island DigitalCommons@URI Infectious Diseases in Corrections Report (IDCR) 2003 HEPP Report: Infectious Diseases in Corrections, Vol. 6 No. 6 HIV & Hepatitis Education Prison Project Follow this and additional works at: http://digitalcommons.uri.edu/idcr Recommended Citation HIV & Hepatitis Education Prison Project, "HEPP Report: Infectious Diseases in Corrections, Vol. 6 No. 6" (2003). Infectious Diseases in Corrections Report (IDCR). Paper 46. http://digitalcommons.uri.edu/idcr/46 This Article is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Infectious Diseases in Corrections Report (IDCR) by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. HIV & HEPATITIS EDUCATION PRISON HEPJune 2003 Vol. 6, Issue 6 P REPORT PROJECT Infectious Diseases in Corrections SPONSOREDBYTHEBROWNMEDICALSCHOOLOFFICEOFCONTINUINGMEDICALEDUCATION. ABOUT HEPP Long-Term Toxicities Associated with HIV and HEPP Report, a forum for Antiretroviral Therapy correctional problem solving, targets correctional physicians, nurses, By Peter J. Piliero, M.D.*, Associate Professor of Medicine, Albany Medical College administrators, outreach workers, and Soon after the introduction of the first antiretroviraldine (3TC) have also been associated with pan- case managers. Published monthly (ARV) agent, zidovudine (AZT), drug-related toxi-creatitis. There may be an added potential for pan- and distributed by email and fax, cities became recognized and well-characterized.creatitis when using combinations of these nucle- HEPP Report provides up-to-the Things have since become more complicated;oside reverse transcriptase inhibitors (NRTIs). moment information HIV/AIDS, there are now 17 ARV agents in four distinct class-Importantly, the concomitant use of alcohol hepatitis, and other infectious es.