Taxonomic Composition and Functional Potential of Sediment Microbial Communities Vary Between Floodplain Wetlands with Differing Management Histories
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -
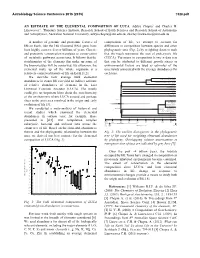
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Pan-Genome Analysis and Ancestral State Reconstruction Of
www.nature.com/scientificreports OPEN Pan‑genome analysis and ancestral state reconstruction of class halobacteria: probability of a new super‑order Sonam Gaba1,2, Abha Kumari2, Marnix Medema 3 & Rajeev Kaushik1* Halobacteria, a class of Euryarchaeota are extremely halophilic archaea that can adapt to a wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl. It consists of the orders: Halobacteriales, Haloferaciales and Natriabales. Pan‑genome analysis of class Halobacteria was done to explore the core (300) and variable components (Softcore: 998, Cloud:36531, Shell:11784). The core component revealed genes of replication, transcription, translation and repair, whereas the variable component had a major portion of environmental information processing. The pan‑gene matrix was mapped onto the core‑gene tree to fnd the ancestral (44.8%) and derived genes (55.1%) of the Last Common Ancestor of Halobacteria. A High percentage of derived genes along with presence of transformation and conjugation genes indicate the occurrence of horizontal gene transfer during the evolution of Halobacteria. A Core and pan‑gene tree were also constructed to infer a phylogeny which implicated on the new super‑order comprising of Natrialbales and Halobacteriales. Halobacteria1,2 is a class of phylum Euryarchaeota3 consisting of extremely halophilic archaea found till date and contains three orders namely Halobacteriales4,5 Haloferacales5 and Natrialbales5. Tese microorganisms are able to dwell at wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl6. Halobacteria, as the name suggests were once considered a part of a domain "Bacteria" but with the discovery of the third domain "Archaea" by Carl Woese et al.7, it became part of Archaea. -

Variations in the Two Last Steps of the Purine Biosynthetic Pathway in Prokaryotes
GBE Different Ways of Doing the Same: Variations in the Two Last Steps of the Purine Biosynthetic Pathway in Prokaryotes Dennifier Costa Brandao~ Cruz1, Lenon Lima Santana1, Alexandre Siqueira Guedes2, Jorge Teodoro de Souza3,*, and Phellippe Arthur Santos Marbach1,* 1CCAAB, Biological Sciences, Recoˆ ncavo da Bahia Federal University, Cruz das Almas, Bahia, Brazil 2Agronomy School, Federal University of Goias, Goiania,^ Goias, Brazil 3 Department of Phytopathology, Federal University of Lavras, Minas Gerais, Brazil Downloaded from https://academic.oup.com/gbe/article/11/4/1235/5345563 by guest on 27 September 2021 *Corresponding authors: E-mails: [email protected]fla.br; [email protected]. Accepted: February 16, 2019 Abstract The last two steps of the purine biosynthetic pathway may be catalyzed by different enzymes in prokaryotes. The genes that encode these enzymes include homologs of purH, purP, purO and those encoding the AICARFT and IMPCH domains of PurH, here named purV and purJ, respectively. In Bacteria, these reactions are mainly catalyzed by the domains AICARFT and IMPCH of PurH. In Archaea, these reactions may be carried out by PurH and also by PurP and PurO, both considered signatures of this domain and analogous to the AICARFT and IMPCH domains of PurH, respectively. These genes were searched for in 1,403 completely sequenced prokaryotic genomes publicly available. Our analyses revealed taxonomic patterns for the distribution of these genes and anticorrelations in their occurrence. The analyses of bacterial genomes revealed the existence of genes coding for PurV, PurJ, and PurO, which may no longer be considered signatures of the domain Archaea. Although highly divergent, the PurOs of Archaea and Bacteria show a high level of conservation in the amino acids of the active sites of the protein, allowing us to infer that these enzymes are analogs. -

Systema Naturae. the Classification of Living Organisms
Systema Naturae. The classification of living organisms. c Alexey B. Shipunov v. 5.601 (June 26, 2007) Preface Most of researches agree that kingdom-level classification of living things needs the special rules and principles. Two approaches are possible: (a) tree- based, Hennigian approach will look for main dichotomies inside so-called “Tree of Life”; and (b) space-based, Linnaean approach will look for the key differences inside “Natural System” multidimensional “cloud”. Despite of clear advantages of tree-like approach (easy to develop rules and algorithms; trees are self-explaining), in many cases the space-based approach is still prefer- able, because it let us to summarize any kinds of taxonomically related da- ta and to compare different classifications quite easily. This approach also lead us to four-kingdom classification, but with different groups: Monera, Protista, Vegetabilia and Animalia, which represent different steps of in- creased complexity of living things, from simple prokaryotic cell to compound Nature Precedings : doi:10.1038/npre.2007.241.2 Posted 16 Aug 2007 eukaryotic cell and further to tissue/organ cell systems. The classification Only recent taxa. Viruses are not included. Abbreviations: incertae sedis (i.s.); pro parte (p.p.); sensu lato (s.l.); sedis mutabilis (sed.m.); sedis possi- bilis (sed.poss.); sensu stricto (s.str.); status mutabilis (stat.m.); quotes for “environmental” groups; asterisk for paraphyletic* taxa. 1 Regnum Monera Superphylum Archebacteria Phylum 1. Archebacteria Classis 1(1). Euryarcheota 1 2(2). Nanoarchaeota 3(3). Crenarchaeota 2 Superphylum Bacteria 3 Phylum 2. Firmicutes 4 Classis 1(4). Thermotogae sed.m. 2(5). -

Mean and SD Archaea Bacteria Genome Size (Mb)
Distributions for kingdom level Primer score mean and SD Genomic GC (%) mean and SD 60 3 2 50 1 Primer score 40 0 Genomic GC (%) Bacteria Bacteria Archaea Archaea kingdom kingdom 16S GC (%) mean and SD Genome size (Mb) mean and SD 65 5 60 4 55 3 16S GC (%) 2 Genome size (Mb) Genome size 50 Bacteria Bacteria Archaea Archaea kingdom kingdom 16S GC (%) Primer score 45 50 55 60 65 0 1 2 3 4 Acidobacteria Acidobacteria Actinobacteria Actinobacteria Aquificae Aquificae Bacteroidetes Bacteroidetes Caldiserica Caldiserica Chlamydiae Chlamydiae Chlorobi Primer scoremeanandSD Chlorobi 16S GC(%)meanandSD Chloroflexi Chloroflexi Chrysiogenetes Chrysiogenetes Crenarchaeota Crenarchaeota Cyanobacteria Cyanobacteria Deferribacteres Deferribacteres Deinococcus−Thermus Deinococcus−Thermus Dictyoglomi Dictyoglomi Elusimicrobia Elusimicrobia phylum phylum Euryarchaeota Euryarchaeota Fibrobacteres Fibrobacteres Firmicutes Firmicutes Fusobacteria Fusobacteria Gemmatimonadetes Gemmatimonadetes Ignavibacteria Ignavibacteria Ignavibacteriae Ignavibacteriae Korarchaeota Korarchaeota Lentisphaerae Lentisphaerae Nanoarchaeota Nanoarchaeota Nitrospirae Nitrospirae Planctomycetes Planctomycetes level phylum for Distributions Proteobacteria Proteobacteria Spirochaetes Spirochaetes Synergistetes Synergistetes Tenericutes Tenericutes Thaumarchaeota Thaumarchaeota Thermodesulfobacteria Thermodesulfobacteria Thermotogae Thermotogae unclassified unclassified Verrucomicrobia Verrucomicrobia Genome size (Mb) Genomic GC (%) 10.0 30 40 50 60 70 2.5 5.0 7.5 Acidobacteria Acidobacteria -

Diversity, Ecology and Evolution of Archaea
REVIEW ARTICLE https://doi.org/10.1038/s41564-020-0715-z Diversity, ecology and evolution of Archaea Brett J. Baker 1 ✉ , Valerie De Anda1, Kiley W. Seitz1, Nina Dombrowski 1, Alyson E. Santoro2 and Karen G. Lloyd 3 Compared to bacteria, our knowledge of archaeal biology is limited. Historically, microbiologists have mostly relied on cultur- ing and single-gene diversity surveys to understand Archaea in nature. However, only six of the 27 currently proposed archaeal phyla have cultured representatives. Advances in genomic sequencing and computational approaches are revolutionizing our understanding of Archaea. The recovery of genomes belonging to uncultured groups from the environment has resulted in the description of several new phyla, many of which are globally distributed and are among the predominant organisms on the planet. In this Review, we discuss how these genomes, together with long-term enrichment studies and elegant in situ measurements, are providing insights into the metabolic capabilities of the Archaea. We also debate how such studies reveal how important Archaea are in mediating an array of ecological processes, including global carbon and nutrient cycles, and how this increase in archaeal diversity has expanded our view of the tree of life and early archaeal evolution, and has provided new insights into the origin of eukaryotes. nitially, all single-celled, non-eukaryotic microorganisms were 16S rRNA gene sequencing revealed many deeply branching classified as the ‘Prokaryota’1 and, later, the ‘Monera’2. These groups19. This uncultivated diversity is commonly referred to as Iearly classifications lumped Bacteria and Archaea into a single ‘microbial dark matter’. group based primarily on morphology. -

Variability in Microbial Community Composition and Function Between Different Niches Within a Coral Reef
Variability in Microbial Community Composition and Function Between Different Niches Within a Coral Reef The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Tout, Jessica, Thomas C. Jeffries, Nicole S. Webster, Roman Stocker, Peter J. Ralph, and Justin R. Seymour. “Variability in Microbial Community Composition and Function Between Different Niches Within a Coral Reef.” Microbial Ecology 67, no. 3 (January 30, 2014): 540–552. As Published http://dx.doi.org/10.1007/s00248-013-0362-5 Publisher Springer US Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/106835 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ Microb Ecol (2014) 67:540–552 DOI 10.1007/s00248-013-0362-5 MICROBIOLOGY OF AQUATIC SYSTEMS Variability in Microbial Community Composition and Function Between Different Niches Within a Coral Reef Jessica Tout & Thomas C. Jeffries & Nicole S. Webster & Roman Stocker & Peter J. Ralph & Justin R. Seymour Received: 1 July 2013 /Accepted: 26 December 2013 /Published online: 30 January 2014 # Springer Science+Business Media New York 2014 Abstract To explore how microbial community composition seawater metagenomes had an enhanced frequency of genes and function varies within a coral reef ecosystem, we per- associated with dynamic processes including motility and formed metagenomic sequencing of seawater from four niches chemotaxis, regulation and cell signalling. -

WHAT CAN LIFE on EARTH TELL US ABOUT LIFE in the UNIVERSE? CHARLES H. LINEWEAVER & ADITYA CHOPRA Planetary Science Institut
J. Seckbach (ed.), Genesis - In The Beginning: Precursors of Life, Chemical Models and Early Biological Evolution, Cellular Origin, Life in Extreme Habitats and Astrobiology 22, 799–815 DOI 10.1007/978-94-007-2941-4_40, ISBN 978-94-007-2940-7.© Springer 2012 WHAT CAN LIFE ON EARTH TELL US ABOUT LIFE IN THE UNIVERSE? CHARLES H. LINEWEAVER & ADITYA CHOPRA Planetary Science Institute, Research School of Astronomy and Astrophysics and the Research School of Earth Sciences, Australian National University, Canberra, Australia We review the most fundamental features common to all terrestrial life. We argue that the ubiquity of these features makes them the best candidates for being features of extraterrestrial life. Other frequently espoused candidates are less secure because they are based on subjective notions of universal fitness, not on features common to all terrestrial life. For example, major transitions in the evolutionary pathway that led to Homo sapiens are sometimes considered to be fundamental transitions in the evolution of all life. However, these “major transitions” are largely arbitrary because a series of different major transitions can be identified along the evolutionary pathway to any extant species. 1. Quirkometry and Terrestrial Life as a Model Organism The universe is filled with stars similar to our Sun (Robles et al., 2008), rocky planets similar to our Earth (Lineweaver and Grether, 2003; Ida and Lin 2004; Mordasini et al., 2009), water like our oceans (Kuchner, 2003; Léger et al., 2004), amino acids like our proteins and all the other ingredients for life (Pizzarello, 2007). But is the universe filled with life? If it is, what kind of life is it? We argue that if there is life out there at all, its basic features are likely to be a subset of the features common to all terrestrial life. -

The Metacyc Database of Metabolic Pathways and Enzymes - a 2019 Update Ron Caspi*, Richard Billington, Ingrid M
Published online 5 October 2019 Nucleic Acids Research, 2020, Vol. 48, Database issue D445–D453 doi: 10.1093/nar/gkz862 The MetaCyc database of metabolic pathways and enzymes - a 2019 update Ron Caspi*, Richard Billington, Ingrid M. Keseler, Anamika Kothari, Markus Krummenacker, Peter E. Midford , Wai Kit Ong, Suzanne Paley , Pallavi Subhraveti and Peter D. Karp* SRI International, 333 Ravenswood Ave, Menlo Park, CA 94025, USA Received September 10, 2019; Revised September 19, 2019; Editorial Decision September 20, 2019; Accepted October 01, 2019 ABSTRACT in the scientific literature (2), and cover both small molecule metabolism and macromolecular metabolism (e.g. protein MetaCyc (MetaCyc.org) is a comprehensive refer- modification). Figure 1 shows an example of a MetaCyc ence database of metabolic pathways and enzymes pathway diagram. Due to its exclusively experimentally from all domains of life. It contains 2749 pathways de- determined data, intensive curation, extensive referencing, rived from more than 60 000 publications, making it and user-friendly and highly integrated interface, MetaCyc the largest curated collection of metabolic pathways. is commonly used in various fields, including genome anno- The data in MetaCyc are evidence-based and richly tation, biochemistry, enzymology, metabolomics, genome curated, resulting in an encyclopedic reference tool and metagenome analysis, and metabolic engineering. for metabolism. MetaCyc is also used as a knowledge In addition to its role as a general reference on base for generating thousands of organism-specific metabolism, MetaCyc is used by the PathoLogic compo- Pathway/Genome Databases (PGDBs), which are nent of the Pathway Tools software (3,4) as a reference database, enabling the computational prediction of the available in BioCyc.org and other genomic portals. -

Systematic Bacteriology Second Edition
BERGEY'S MANUAL® OF Systematic Bacteriology Second Edition Volume One The Archaea and the Deeply Branching and Phototrophic Bacteria Springer New York Berlin Heidelberg Barcelona HongKong London Milan Paris Singapore Tokyo BERGEY'S MANUAL® OF Systematic Bacteriology Second Edition Volume One The Archaea and the Deeply Branching and Phototrophic Bacteria David R. Boone Richard W. Castenholz EDITORS, VOLUME ONE George M. Garrity EDITOR-IN-CHIEF EDITORIAL BOARD James T. Staley, Chairman, David R. Boone, Vice Chairman, Don J. Brenner, Richard W. Castenholz, George M. Garrity, Michael Goodfellow, Noel R. Krieg, Fred A. Rainey, Karl-Heinz Schleifer WITH CONTRIBUTIONS FROM 105 COLLEAGUES Springer George M. Garrity Department of Microbiology and Molecular Genetics Bergey's Manual Trust Michigan State University East Lansing, MI 48824-1101 USA Library of Congress Cataloging-in-Publication Data Bergey's manual of systematic bacteriology / David R. Boone, Richard W. Castenholz, editors, volume 1 ; George M. Garrity, editor-in-chief.-2nd ed. p.cm. Includes bibliographical references and index. Contents: v.I. The archaea and the deeply branching and phototrophic bacteria. ISBN 0-387-98771-1 (alk. paper) 1. Bacteria-Classification. I. Title: Systematic bacteriology. II. Boone, David R. III. Castenholz, Richard W. IV. Garrity, George M. QR81.B462001 579.3'01 '2-dc21 2001020400 With 330 illustrations The following proprietary names of products are used in this volume: Casamino@ acids; Vector NTI®; XLI0-Gold®; Gelrite®; Tryptone@l; Phytagel@; bio-Trypcasew; Trypticasev; Oxoid® purified agar. Printed on acid-free paper. First edition published 1984-1989 by Bergey's Manual Trust and Williams & Wilkins, Baltimore. © 2001 Bergey's Manual Trust All rights reserved.