Implications of BRCA1 Mutations in Basal-Like Breast Cancer Development and Treatment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
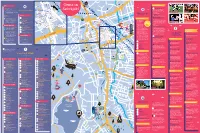
Gems in Seinäjoki
4 <- Vaasa 34 Y Valtionkatu stie 3 ga X Ä 10 en CULTURE, MUSEUMS INFO R Puskantie Gems in C 59 Kokkola -> Vaasantie 6 Emergency 50 A3 Seinäjoki City Theatre Koskenalantie 18 Z Awarded as the 2015 Theater of On call between 8am–8pm, Accommodation CITY CENTER 28 T Seinäjoki the Year. Versatile repertoire. Also p. +358 (0)6 425 5311 Koulukatu 6 58 39 has a lunch restaurant in gorgeous At nighttime between 8pm–8am, 33 surroundings. SJK SEINÄJOEN PALLOKERHO PAUKANEVA p. +358 (0)6 415 4555 22 70 Maamiehenk. Alvar Aallon katu 22 30 72 12 53 62 What to do Keskuskatu -> 37 Seinäjoki City Orchestra Police HOTELS 5 Juhonkatu 4 18 16 <- Kauhajoki Follow the repertoire at www.skor.fi 71 66 49 31 24 Sammonkatu 64 3 D Provincial museum of Tourist information 1 Scandic Seinäjoki <- Kalevankatu55 1 21 19 South Ostrobothnia South Ostrobothnia Tourist Kauppakatu 10 69 AALTO-CENTRE Porvarink. 56 A versatile and extensive museum Service Ltd. Located in the 13 15 6014 67 POHJA 2 Hotel Fooninki area located in the beautiful park area Travel Center (Matkakeskus) 61 1 Verkatehtaankatu 40 DAP The administrative Kaarretie 4 19 52 Ruukintie of Törnävä. The permanent exhibition premises 50 1 Valtionkatu and cultural center SQUASH & BOWLING CENTER THE DUDESONS ACTIVITY PARK 42 in the stone barn is about the past p. +358 (0)6 420 9090 3 Hotel-Restaurant Alma Kutojankatu of Seinäjoki is F 29 and present of Seinäjoki and the Ruukintie 4 20 Kauppakatu 67-> 48 one the most 32 57 Kulmak. substantial works province. -

Citycon Annual Report 2007 Annual Report Citycon Business and Property Portfolio
Pohjoisesplanadi 35 AB Tel. +358 9 680 36 70 www.citycon.fi FI-00100 Helsinki, Fax +358 9 680 36 788 [email protected] Finland Annual Report 2007 Contents 2007 Citycon in Brief .....................................................................................................................................1 Citycon as an Investment and Information for Shareholders .............................2 CEO’s Review ...........................................................................................................................................4 Business Environment .....................................................................................................................6 Citycon Annual Report Annual Report Citycon Business and Property Portfolio ...............................................................................................9 Finland ....................................................................................................................................................27 Sweden ...................................................................................................................................................30 Baltic Countries ................................................................................................................................32 Human Resources ...........................................................................................................................34 Profit Performance and Financial Position ..................................................................37 -

Q1 Presentation Helsinki 26 April, 2007 Disclaimer
Q1 Presentation Helsinki 26 April, 2007 Disclaimer This document and the information contained herein is strictly confidential and is being provided to you solely for your information. This document may not be retained by you and neither this document nor the information contained herein may be reproduced, further distributed to any other person or published, in whole or in part, for any purpose. These materials do not constitute an offer or solicitation of an offer to buy securities anywhere in the world. No securities of Citycon Oyj (the “Company”) have been or will be registered under the U.S. Securities Act of 1933, as amended (the “Securities Act”). Certain ordinary shares of the Company have been offered to “qualified institutional buyers” (as such term is defined in Rule 144A (“Rule 144A”)) under the Securities Act, in transactions not involving a public offering within the meaning of the Securities Act. Accordingly, such shares are “restricted securities” within the meaning of Rule 144 and may not be resold or transferred in the United States, absent an exemption from SEC registration or an effective registration statement. There will be no public offering of the securities in the United States. Subject to certain exceptions, neither this document nor any part or copy of it may be taken or transmitted into the United States or distributed, directly or indirectly, in the United States, or to any “U.S. Person” as that term is defined in Regulation S under the Securities Act. Neither this document nor any part or copy of it may be taken or transmitted into Australia, Canada or Japan, or distributed directly or indirectly in Canada or distributed or redistributed in Japan or to any resident thereof. -

Public Value and Performance of Libraries
Qualitative and Quantitative Methods in Libraries (QQML) 6: 271-281, 2017 Public value and performance of libraries Antti Seppänen1, Markku Laitinen2, Tomi Rajala3 Harri Laihonen3 1 Regional State Administrative Agency of Southern Finland 2 National Library of Finland 3 University of Tampere, Faculty of Management Abstract: This paper addresses the changing role of libraries as central knowledge hubs of an information society. It questions the relevance of the existing performance measures and asks what kind of public value we are expecting from the libraries. The paper illustrates its main arguments with the empirical context of the Finland and shortly introduces the case of ‘Iso Omena’ in Espoo, Finland where a novel approach for providing public value has been implemented. The paper develops two arguments to guide the future research in the area. First, new indicators and measurement practices are needed if we want to evaluate more comprehensively the public value of libraries. Second, performance management and evaluation taking place in libraries calls for active performance dialogue between the relevant interest groups in order to determine the public value of libraries and performance measures tracking how this value is produced. Keywords: library, public value, performance, management control, organizational learning 1. Introduction Libraries role as corner stone of information society is important, but the world is changing and therefore also libraries need to rethink their role and value proposition. How can libraries today and tomorrow create most value to customers and taxpayers? One way to seek answers to this question is a public value approach as a set of normative values that create a foundation for public activities such as libraries is this case. -

Tampere Travel and Service Centre
TAMPERE TRAVEL AND SERVICE CENTRE ASSESSMENT OF THE FINANCIAL CONDITIONS FOR IMPLEMENTATION AND THE COMMERCIAL CONCEPT 9 October 2015 NEWSEC VALUATION OY Contents 1 Background ........................................................................................................... 3 2 The scope .............................................................................................................. 3 3 The Travel and Service Centre – phasing and scheduling of implementation .................................................................................................... 4 3.1 Projects in central Tampere .......................................................................................... 5 3.2 Construction of parking spaces outside the Travel and Service Centre area ..... 11 3.3 Summary ........................................................................................................................ 12 4 Assessment of the financial conditions for the project ................................ 15 4.1 The property investment market in Tampere ........................................................... 15 4.2 Estimated unit costs and values ................................................................................. 16 4.3 Financial assessment .................................................................................................. 18 5 Market position of the Travel and Service Centre .......................................... 19 5.1 Commercial centres in Tampere ............................................................................... -

Best Budget Accommodations in Tampere"
"Best Budget Accommodations in Tampere" Erstellt von : Cityseeker 6 Vorgemerkte Orte Hotelli Cumulus Koskikatu "On the Banks of Tammerkoski" Located on the banks of Tammerkoski Rapids, Hotel Cumulus Koskikatu is in the heart of Tampere. You may choose any direction from the hotel and be certain of ending up very soon at a number of attractions. Koskikeskus shopping center, Stockmann department store, the railway station and the Central Square can practically be seen from the hotel. One of the main by Booking.com nightclubs in Tampere, Night Life, popular among the young and youthful adults, is next door to the hotel. +358 3 242 41 11 www.cumulus.fi/hotellit/k [email protected] Koskikatu 5, Tampere oskikatu/en_GB/koskikatu/ i Sokos Hotel Ilves "The best quality in town" Located on the banks of Tammerkoski Rapids, in the very center of the city, Hotel Ilves rises well above any other building nearby. It represents the highest quality in Tampere and is particularly popular among business travelers. Many services lie close by, like Koskikeskus shopping center. Tampere tourist office, Tampere Theatre, the railway station and a number by Booking.com of department stores, boutiques and restaurants are within a few hundred metres. Ilves is also famous for its nightclub. +358 3 262 62 62 [email protected] Hatanpään valtatie 1, Tampere Scandic Hotel Tampere "Style In the Centre" Scandic Hotel Tampere is a modern urban hotel, centrally located right next to the railway station. A wide variety of city center services, as well as Tampere's versatile cultural offerings are within walking distance. -

Finland's Largest Entertainment Centre to Open in Vantaa
ISSUE 33 (65) • 29 AUGUST – 4 SEPTEMBER 2008 • €3 • WWW.HELSINKITIMES.FI DOMESTIC NEWS BUSINESS SPORT LIFESTYLE CULTURE Dead Forestry HJK breaks Peace The world zones in compared Inter’s is in of art Baltic Sea in EU-27 undefeated run the air exchange page 5 page 10 page 13 page 14 page 15 Violence among pupils has increased at an alarming rate HEIDI LEHTONEN - HT early intervention are central to MICHAEL NAGLER – HT decreasing school bullying and vi- olence. It is important to increase BULLYING has become more com- knowledge about how we perceive mon in comprehensive schools. Sev- violence and what it means to youth, en per cent of girls and one tenth of parents and those who work among boys said that they are bullied at least young people,” Pietikäinen says. once a week. A fi fth of junior high The results of the Kiva Koulu school students on average have ex- (Nice school) programme, which was perienced physical threats during the developed at the University of Turku last year. Fights and threatening with with funding from the Ministry of violence have increased considerably Education, have appeared prom- among boys in comprehensive school. ising. In the experimental schools These results are revealed in a school which ran for a year, the proportion health survey conducted by STAKES, of children who were repeatedly bul- the National Research and Develop- lied as well as those who bullied oth- ment Centre for Welfare and Health. ers fell by about 40 per cent. Over 100,000 comprehensive school, Elisa Poskiparta of the Universi- vocational school and high school stu- ty of Turku, a senior researcher of the dents took part in the survey. -

Pushing the City Forward with €1 Billion of Investments
Pushing the city forward with €1 billion of investments WE HAVE SPACE FOR ALL THE GOOD THINGS 2 Seinäjoki PushingMiljardilla the enemmän city forward elinvoimaa with €1 billion of investments DIY Seinäjoki has never shied away from building BIG. “Too outlandish” is not really part of the local vocabulary. Once work gets underway, there is no brake pedal. The Aalto Centre – an internationally 1960 renowned architectural destination Kyrkösjärvi – we did not have a lake, 1980 so we made one Jouppilanvuori (118 m) – we could not find 1980 a mountain, so we made one, and it became the city’s highest point Frami – the heart of the city’s business 2000 and science park. 46,500 m², 3,000 students, 80 companies, 1,000 experts Apila – A library building of 2012 unrivalled beauty OmaSP Stadium – Finland’s classiest 2016 football stadium Boosting the city’s vitality with 2017– investments of €1 billion 2021 Pushing the city forward with €1 billion of investments Seinäjoki 3 Jorma Rasinmäki Billion Mayor Space for pace success The pace of private investment in Seinäjoki is soaring beautifully. Half of the expected billion "THE RATE OF APARTMENT construction in the capital of space is one of the briskest in euros of investment has already been realised, Finland. Here, the number of workplaces grows, while many new investments are in the pipeline. while unemployment continues to fall. Seinäjoki has often been chosen as Finland’s - WE PUT TOGETHER A LIST of planned private sector investments in best partner for companies. We are in the cent- Seinäjoki from the start of 2017 to 2021. -

Citycon Annual Report 2006
Annual Report ’06 Contents Citycon in Brief ............................................................................................................1 Citycon as an Investment and Information for Shareholders ....2 Mission, Vision, Goals and Strategy ...............................................................4 CEO’s Review ..................................................................................................................6 Business Environment ............................................................................................8 Business and Property Portfolio ...................................................................10 Finland ............................................................................................................................20 Sweden ...........................................................................................................................24 Baltic Countries ........................................................................................................28 Human Resources ...................................................................................................30 Profit Performance and Financial Position ...........................................32 Risks and Risk Management ...........................................................................36 Corporate Governance .........................................................................................38 Glossary ANNUAL REPORT 2006 CITYCON OYJ 1 Forward-Looking Statements Some statements -

Citycon Presentations
Citycon's Capital Markets Day 22 September 2010 Tallinn, Estonia Strategy Citycon • wants to be the leading shopping centre owner, operator and developer in the Nordic and Baltic countries. • invests in shopping centres and retail properties in major growing cities with good demographics. • seeks growth through matching acquisitions and property development. • adds value on investment across the portfolio by professional active management produced by in-house strong personnel. • operates by high sustainability standards. • seeks actively joint-venture arrangements with high-class investors and manages investment on their behalf. • has a strong balance sheet with competitive and well diversified funding sources and low financial risk exposures. 2 Q2 2010 Citycon's Financial Position CFO, Executive Vice President Eero Sihvonen Main Points Q2 2010 Q2 2010 (vs. Q1 2010) • The fair value change was EUR 22.9 million (EUR 0.8 m) and the market value of property portfolio was EUR 2,229.5 million (EUR 2,193.5 m) • The valuation yield 6.6% (6.6%) by external appraiser • Net rental income was EUR 31.8 million (EUR 30.6m) • Direct result per share (diluted EPRA EPS) EUR 0.05 (EUR 0.05) Q1-Q2 2010 (vs. Q1-Q2 2009) • Turnover increased by 7.3% to EUR 98.1 million (EUR 91.5m) • Direct result per share (EPRA EPS) EUR 0.10 (EUR 0.11) • Direct result was EUR 21.5 million (EUR 24.2 m) • Net cash from operating activities per share EUR 0.05 (EUR 0.19) due to extraordinary and timing items. • Profit before taxes EUR 52.0 million (EUR -28.7 m), incl. -

Five-Star City Centre 2017-2030
FIVE-STAR CITY CENTRE 2017-2030 TAMPERE CITY CENTRE DEVELOPMENT PROGRAMME 2017-2030 CITY BOARD 18 APRIL 2017 FIVE-STAR CITY CENTRE 2017-2030 NEW ∠ A DECK ACROSS THE RAILWAY ZONE AND NEW, EFFICIENT CONSTRUCTION ∠ NEW RESIDENTIAL AREAS ON THE SHORES OF LAKE NÄSIJÄRVI AND LAKE PYHÄJÄRVI ∠ INFILL DEVELOPMENT OF OLD BUSINESS AND HOUSING BLOCKS ∠ MORE PLEASANT MARKET PLACES, SQUARES, YARDS AND STREETS ∠ SHORE ROUTES HAVE BEEN CREATED AND THE CITY SHORES ARE USED BY PEOPLE ∠ THE TRAMWAY, THE TRAVEL CENTRE, THE P-KUNKKU PARKING FACILITY, NÄSINKALLIO JUNCTION VISION ∠ PART OF A GOOD CITY THAT IS WELCOMING TO ALL ∠ A LEADER IN THE BUSINESS AND INDUSTRIAL SECTOR ∠ SMOOTH AND ACCESSIBLE ∠ EVENTS, EXPERIENCES AND SERVICES ∠ LOCAL AND INTERNATIONAL OBJECTIVES ∠ NEW RESIDENTS +15,000 ∠ NEW JOBS +15,000 ∠ AN INCREASING VITALITY AWARD THE ELÄVÄT KAUPUNKIKESKUSTAT ASSOCIATION 2016: THE BEST CITY CENTRE IN FINLAND www.tampere.fi/keskustahanke > Tampereen keskustan kehittämisohjelma ImagImaImImmamagagageCe::C: C ityiitttyy ofoffT Tamampeampampempmpeperere,re,e, cityccitttyy modmomooddeellvl viisuaisuissusuasuaualillisaisaiisssaattiotiontiionioioonn bybbMyyM M YArYAY ArA chitchichhithihititeectsectctsccttts,,2 220011717.7 4 3 5 12 7 14 13 12 10 10 14 13 12 12 14 7 13 6 1 8 11 9 2 Oblique image: Lentokuva Vallas Oy, 20166. FIVE-STAR CITY CENTRE 2017-2030 KEY PROJECTS 2017 1. TRAVEL AND SERVICE CENTRE 9. TULLI+ 2. DECK AND THE ARENA 10. BUSINESS BLOCKS 3. SÄRKÄNNIEMI 11. RATINA 4. EVENTS 12. SQUARES AND MARKET 5. RANTA-TAMPELLA PLACES IN THE CITY CENTRE 6. ETELÄPUISTO 13. EPICENTRE 7. TAMMELA AND AMURI 14. HÄMEENKATU STREET 8. TAMMELA STADIUM NEW 3D GALLERY OF TAMPERE THE 3D GALLERY OF TAMPERE, WHICH WAS TAKEN INTO USE IN EARLY 2017, FUNCTIONS AS A TOOL FOR CITY PLANNING, CONSTRUCTION PLANNING, STREET AND ENVIRONMENTAL PLANNING, AS WELL AS FOR THE ILLUSTRATIONS AND DIALOGUE IN THESE FIELDS. -

Finnish Studies
JOURNAL OF INNISH TUDIES F S From Cultural Knowledge to Cultural Heritage: Finnish Archives and Their Reflections of the People Guest Editors Pia Olsson and Eija Stark Theme Issue of the Journal of Finnish Studies Volume 18 Number 1 October 2014 ISSN 1206-6516 ISBN 978-1-937875-96-1 JOURNAL OF FINNISH STUDIES EDITORIAL AND BUSINESS OFFICE Journal of Finnish Studies, Department of English, 1901 University Avenue, Evans 458 (P.O. Box 2146), Sam Houston State University, Huntsville, TX 77341-2146, USA Tel. 1.936.294.1402; Fax 1.936.294.1408 SUBSCRIPTIONS, ADVERTISING, AND INQUIRIES Contact Business Office (see above & below). EDITORIAL STAFF Helena Halmari, Editor-in-Chief, Sam Houston State University; [email protected] Hanna Snellman, Co-Editor, University of Helsinki; [email protected] Scott Kaukonen, Assoc. Editor, Sam Houston State University; [email protected] Hilary Joy Virtanen, Asst. Editor, Finlandia University; hilary.virtanen@finlandia. edu Sheila Embleton, Book Review Editor, York University; [email protected] EDITORIAL BOARD Börje Vähämäki, Founding Editor, JoFS, Professor Emeritus, University of Toronto Raimo Anttila, Professor Emeritus, University of California, Los Angeles Michael Branch, Professor Emeritus, University of London Thomas DuBois, Professor, University of Wisconsin Sheila Embleton, Distinguished Research Professor, York University Aili Flint, Emerita Senior Lecturer, Associate Research Scholar, Columbia University Richard Impola, Professor Emeritus, New Paltz, New York Daniel Karvonen, Senior Lecturer, University of Minnesota, Minneapolis Andrew Nestingen, Associate Professor, University of Washington, Seattle Jyrki Nummi, Professor, Department of Finnish Literature, University of Helsinki Juha Pentikäinen, Professor, Institute for Northern Culture, University of Lapland Douglas Robinson, Professor, Dean, Hong Kong Baptist University Oiva Saarinen, Professor Emeritus, Laurentian University, Sudbury George Schoolfield, Professor Emeritus, Yale University Beth L.