Hereditary Leiomyomatosis and Renal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Skin and Breast Disease in the Differential Diagnosis of Chest Pain
Skin and breast disease in the differential diagnosis of chest pain Author Muir, Jim, Yelland, Michael Published 2010 Journal Title Medical Clinics of North America DOI https://doi.org/10.1016/j.mcna.2010.01.006 Copyright Statement © 2010 Elsevier. This is the author-manuscript version of this paper. Reproduced in accordance with the copyright policy of the publisher. Please refer to the journal's website for access to the definitive, published version. Downloaded from http://hdl.handle.net/10072/33712 Griffith Research Online https://research-repository.griffith.edu.au ARTICLE IN PRESS 1 2 Skin and Breast 3 4 Disease in the 5 6 Differential 7 8 Diagnosis of 9 10 Chest Pain 11 12 a, b ½Q2½ Q3 Jim Muir *, Michael Yelland ½Q4½ Q5 KEYWORDS 13 14 Chest pain Skin diseases Herpes zoster PROOF 15 Breast Neoplasm 16 17 18 Pain is not a symptom commonly associated with skin disease. This is especially so 19 when considering the known skin problems that have a presenting symptom of chest 20 pain that could potentially be confused with chest pain from other causes. 21 22 PAINFUL SKIN CONDITIONS 23 24 Several extremely painful and tender skin conditions present with dramatic clinical 25 signs. Inflammatory disorders such as pyoderma gangrenosum, skin malignancies, 26 both primary and secondary, acute bacterial infections such as erysipelas or cellulitis, 27 and multiple other infections are commonly extremely painful and tender. As these 28 conditions manifest with obvious skin signs such as swelling, erythema, localized 29 tenderness, fever, lymphangitis, and lymphadenopathy, there is little chance of misdi- 30 agnosis of symptoms as caused by anything other than a cutaneous pathology. -

Benign Tumors and Tumor-Like Lesions of the Vulva
Please do not remove this page Benign Tumors and Tumor-like Lesions of the Vulva Heller, Debra https://scholarship.libraries.rutgers.edu/discovery/delivery/01RUT_INST:ResearchRepository/12643402930004646?l#13643525330004646 Heller, D. (2015). Benign Tumors and Tumor-like Lesions of the Vulva. In Clinical Obstetrics & Gynecology (Vol. 58, Issue 3, pp. 526–535). Rutgers University. https://doi.org/10.7282/T3RN3B2N This work is protected by copyright. You are free to use this resource, with proper attribution, for research and educational purposes. Other uses, such as reproduction or publication, may require the permission of the copyright holder. Downloaded On 2021/09/23 14:56:57 -0400 Heller DS Benign Tumors and Tumor-like lesions of the Vulva Debra S. Heller, MD From the Department of Pathology & Laboratory Medicine, Rutgers-New Jersey Medical School, Newark, NJ Address Correspondence to: Debra S. Heller, MD Dept of Pathology-UH/E158 Rutgers-New Jersey Medical School 185 South Orange Ave Newark, NJ, 07103 Tel 973-972-0751 Fax 973-972-5724 [email protected] Funding: None Disclosures: None 1 Heller DS Abstract: A variety of mass lesions may affect the vulva. These may be non-neoplastic, or represent benign or malignant neoplasms. A review of benign mass lesions and neoplasms of the vulva is presented. Key words: Vulvar neoplasms, vulvar diseases, vulva 2 Heller DS Introduction: A variety of mass lesions may affect the vulva. These may be non-neoplastic, or represent benign or malignant neoplasms. Often an excision is required for both diagnosis and therapy. A review of the more commonly encountered non-neoplastic mass lesions and benign neoplasms of the vulva is presented. -

A Single Case Report of Granular Cell Tumor of the Tongue Successfully Treated Through 445 Nm Diode Laser
healthcare Case Report A Single Case Report of Granular Cell Tumor of the Tongue Successfully Treated through 445 nm Diode Laser Maria Vittoria Viani 1,*, Luigi Corcione 1, Chiara Di Blasio 2, Ronell Bologna-Molina 3 , Paolo Vescovi 1 and Marco Meleti 1 1 Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy; [email protected] (L.C.); [email protected] (P.V.); [email protected] (M.M.) 2 Private practice, Centro Medico Di Blasio, 43121 Parma; Italy; [email protected] 3 Faculty of Dentistry, University of the Republic, 14600 Montevideo, Uruguay; [email protected] * Correspondence: [email protected] Received: 10 June 2020; Accepted: 11 August 2020; Published: 13 August 2020 Abstract: Oral granular cell tumor (GCT) is a relatively rare, benign lesion that can easily be misdiagnosed. Particularly, the presence of pseudoepitheliomatous hyperplasia might, in some cases, lead to the hypothesis of squamous cell carcinoma. Surgical excision is the treatment of choice. Recurrence has been reported in up to 15% of cases treated with conventional surgery. Here, we reported a case of GCT of the tongue in a young female patient, which was successfully treated through 445 nm diode laser excision. Laser surgery might reduce bleeding and postoperative pain and may be associated with more rapid healing. Particularly, the vaporization effect on remnant tissues could eliminate GCT cells on the surgical bed, thus hypothetically leading to a lower rate of recurrence. In the present case, complete healing occurred in 1 week, and no recurrence was observed after 6 months. Laser surgery also allows the possibility to obtain second intention healing. -

Atypical Fibroids
Hereditary leiomyomatosis and renal cell cancer: Cutaneous lesions & atypical fibroids The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Bortoletto, Pietro, Jennifer L. Lindsey, Liping Yuan, Bradley J. Quade, Antonio R. Gargiulo, Cynthia C. Morton, Elizabeth A. Stewart, and Raymond M. Anchan. 2017. “Hereditary leiomyomatosis and renal cell cancer: Cutaneous lesions & atypical fibroids.” Case Reports in Women's Health 15 (1): 31-34. doi:10.1016/ j.crwh.2017.06.004. http://dx.doi.org/10.1016/j.crwh.2017.06.004. Published Version doi:10.1016/j.crwh.2017.06.004 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:35982080 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Case Reports in Women's Health 15 (2017) 31–34 Contents lists available at ScienceDirect Case Reports in Women's Health journal homepage: www.elsevier.com/locate/crwh Hereditary leiomyomatosis and renal cell cancer: Cutaneous MARK lesions & atypical fibroids Pietro Bortolettoa,b,c,1, Jennifer L. Lindseya,b,1, Liping Yuand, Bradley J. Quadec,d, Antonio R. Gargiuloa,b,c, Cynthia C. Mortonb,c,d,e,f, Elizabeth A. Stewartg, ⁎ Raymond M. Anchana,b,c, a Division of Reproductive Endocrinology and Infertility, Boston, MA, USA b Department of Obstetrics, Gynecology and Reproductive Biology, -
Study to Evaluate the Safety and Effectiveness of Zostavax™ in Subjects 50 - 59 Years of Age (V211-022) - Full Text View - Clinicaltrials.Gov
Study to Evaluate the Safety and Effectiveness of Zostavax™ in Subjects 50 - 59 Years of Age (V211-022) - Full Text View - ClinicalTrials.gov Example: "Heart attack" AND "Los Angeles" Search for studies: A service of the U.S. National Institutes of Health Advanced Search Help Studies by Topic Glossary Find Studies About Clinical Studies Submit Studies Resources About This Site Home Find Studies Search Results Study Record Detail Text Size Trial record 1 of 1 for: NCT00534248 Previous Study | Return to List | Next Study Study to Evaluate the Safety and Effectiveness of Zostavax™ in Subjects 50 - 59 Years of Age (V211-022) This study has been completed. ClinicalTrials.gov Identifier: NCT00534248 Sponsor: Merck Sharp & Dohme Corp. First received: September 21, 2007 Last updated: August 11, 2015 Information provided by (Responsible Party): Last verified: August 2015 Merck Sharp & Dohme Corp. History of Changes Full Text View Tabular View Study Results Disclaimer How to Read a Study Record Purpose This study will look at how well Zostavax™ works in preventing shingles in participants ages 50-59 years old. Condition Intervention Phase Shingles Biological: Zoster Vaccine, Live (Zostavax™) Phase 3 Biological: Comparator: Placebo Study Type: Interventional Study Design: Allocation: Randomized Endpoint Classification: Safety/Efficacy Study Intervention Model: Parallel Assignment Masking: Double Blind (Subject, Investigator) Primary Purpose: Prevention Official Title: A Phase III Clinical Trial to Evaluate the Efficacy, Immunogenicity, Safety and -

Cutaneous Melanoma Metastasizing to Leiomyoma Uteri
CASE REPORT Tumour-to-tumour Metastasis: Cutaneous Melanoma Metastasizing to Leiomyoma Uteri Jelena Amidzic1, Nenad Solajic2, Aleksandra Fejsa Levakov1, Matilda Djolai1 and Nada Vuckovic2 1Department for Histology and Embryology / Pathology2, Centre for Pathology and Histology, Clinical Centre of Vojvodina, Faculty of Medicine, University of Novi Sad, HajdukVeljkova 1-3, 21000 Novi Sad, Serbia ABSTRACT The incidence of melanoma is increasing worldwide. It is known that melanoma frequently progresses to metastatic disease. The aim of this report is to emphasise the metastatic potential of cutaneous melanoma to various body areas, as well as the ability to produce unexpected presentation of the disease. A 48-year female had a myomatous uterus and underwent hysterectomy. At the pathological examination, multiple leiomyomas were diagnosed and in one of them, the metastatic melanoma was found, the later confirmed with immunohistochemical analysis. The medical history revealed that the patient was previously operated two years back due to skin superficial spreading melanoma. The metastasis to uterine leiomyoma was the first site of distant spread. Melanoma is a type of tumour with aggressive and unpredictable behaviour, so metastases to unexpected localisations could occur. A careful examination of patient's body is mandatory, including the remote areas and even benign tumours. Key Words: Tumout-to-tumour metastasis, Melanoma, Leiomyoma. How to cite this article: Amidzic J, Solajic N, Fejsa Levakov A, Djolai M, Vuckovic N. Tumour-to-tumour metastasis: cutaneous melanoma metastasizing to leiomyoma uteri. J Coll Physicians Surg Pak 2019; 29 (Supplement 2):S112-S113. INTRODUCTION Recognised by T-cells (MART1) and polyclonal or 2 Melanoma is an aggressive, highly malignant disease monoclonal S100 protein. -

Inherited Skin Tumour Syndromes
CME GENETICS Clinical Medicine 2017 Vol 17, No 6: 562–7 I n h e r i t e d s k i n t u m o u r s y n d r o m e s A u t h o r s : S a r a h B r o w n , A P a u l B r e n n a n B a n d N e i l R a j a n C This article provides an overview of selected genetic skin con- and upper trunk. 1,2 These lesions are fibrofolliculomas, ditions where multiple inherited cutaneous tumours are a cen- trichodiscomas and acrochordons. Patients are also susceptible tral feature. Skin tumours that arise from skin structures such to the development of renal cell carcinoma, lung cysts and as hair, sweat glands and sebaceous glands are called skin pneumothoraces. 3 appendage tumours. These tumours are uncommon, but can Fibrofolliculomas and trichodiscomas clinically present as ABSTRACT have important implications for patient care. Certain appenda- skin/yellow-white coloured dome shaped papules 2–4 mm in geal tumours, particularly when multiple lesions are seen, may diameter (Fig 1 a and Fig 1 b). 4 These lesions usually develop indicate an underlying genetic condition. These tumours may in the third or fourth decade.4 In the case of fibrofolliculoma, not display clinical features that allow a secure diagnosis to be hair specific differentiation is seen, whereas in the case of made, necessitating biopsy and dermatopathological assess- trichodiscoma, differentiation is to the mesodermal component ment. -

2016 Essentials of Dermatopathology Slide Library Handout Book
2016 Essentials of Dermatopathology Slide Library Handout Book April 8-10, 2016 JW Marriott Houston Downtown Houston, TX USA CASE #01 -- SLIDE #01 Diagnosis: Nodular fasciitis Case Summary: 12 year old male with a rapidly growing temple mass. Present for 4 weeks. Nodular fasciitis is a self-limited pseudosarcomatous proliferation that may cause clinical alarm due to its rapid growth. It is most common in young adults but occurs across a wide age range. This lesion is typically 3-5 cm and composed of bland fibroblasts and myofibroblasts without significant cytologic atypia arranged in a loose storiform pattern with areas of extravasated red blood cells. Mitoses may be numerous, but atypical mitotic figures are absent. Nodular fasciitis is a benign process, and recurrence is very rare (1%). Recent work has shown that the MYH9-USP6 gene fusion is present in approximately 90% of cases, and molecular techniques to show USP6 gene rearrangement may be a helpful ancillary tool in difficult cases or on small biopsy samples. Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors, 5th edition. Mosby Elsevier. 2008. Erickson-Johnson MR, Chou MM, Evers BR, Roth CW, Seys AR, Jin L, Ye Y, Lau AW, Wang X, Oliveira AM. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011 Oct;91(10):1427-33. Amary MF, Ye H, Berisha F, Tirabosco R, Presneau N, Flanagan AM. Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch. 2013 Jul;463(1):97-8. CONTRIBUTED BY KAREN FRITCHIE, MD 1 CASE #02 -- SLIDE #02 Diagnosis: Cellular fibrous histiocytoma Case Summary: 12 year old female with wrist mass. -

Clinical Features and Histological Description of Tongue Lesions in a Large Northern Italian Population
Med Oral Patol Oral Cir Bucal. 2015 Sep 1;20 (5):e560-5. Retrospective study on tongue lesions Journal section: Oral Medicine and Pathology doi:10.4317/medoral.20556 Publication Types: Research http://dx.doi.org/doi:10.4317/medoral.20556 Clinical features and histological description of tongue lesions in a large Northern Italian population Alessio Gambino 1, Mario Carbone 1, Paolo-Giacomo Arduino 1, Marco Carrozzo 2, Davide Conrotto 1, Carlotta Tanteri 1, Lucio Carbone 3, Alessandra Elia 1, Zaira Maragon 3, Roberto Broccoletti 1 1 Department of Surgical Sciences, Oral Medicine Section, CIR - Dental School, University of Turin, Turin, Italy 2 Oral Medicine Department, Centre for Oral Health Research, Newcastle University, Newcastle upon Tyne, UK 3 Private practice, Turin Correspondence: Oral Medicine Section University of Turin CIR – Dental School Gambino A, Carbone M, Arduino PG, Carrozzo M, Conrotto D, Tanteri Via Nizza 230, 10126 C, Carbone L, Elia A, Maragon Z, Broccoletti R. Clinical features and Turin, Italy histological description of tongue lesions in a lar�������������������������ge Northern Italian popu- [email protected] lation. Med Oral Patol Oral Cir Bucal. 2015 Sep 1;20 (5):e560-5. http://www.medicinaoral.com/medoralfree01/v20i5/medoralv20i5p560.pdf Article Number: 20556 http://www.medicinaoral.com/ Received: 21/12/2014 © Medicina Oral S. L. C.I.F. B 96689336 - pISSN 1698-4447 - eISSN: 1698-6946 Accepted: 25/04/2015 eMail: [email protected] Indexed in: Science Citation Index Expanded Journal Citation Reports Index Medicus, MEDLINE, PubMed Scopus, Embase and Emcare Indice Médico Español Abstract Background: Only few studies on tongue lesions considered sizable populations, and contemporary literature does not provide a valid report regarding the epidemiology of tongue lesions within the Italian population. -

Acute Kidney Injury Due to Menstruation-Related Disseminated Intravascular Coagulation in an Adenomyosis Patient: a Case Report
CASE REPORT Nephrology DOI: 10.3346/jkms.2010.25.9.1372 • J Korean Med Sci 2010; 25: 1372-1374 Acute Kidney Injury due to Menstruation-related Disseminated Intravascular Coagulation in an Adenomyosis Patient: A Case Report Jungmin Son1,4, Dong Won Lee1,4, The authors report a case of acute kidney injury (AKI) resulting from menstruation-related Eun Young Seong1,4, Sang Heon Song1,4, disseminated intravascular coagulation (DIC) in an adenomyosis patient. A 40-yr-old Soo Bong Lee1,4, Jin Kang1, woman who had received gonadotropin for ovulation induction therapy presented with Byeong Yun Yang1, Su Jin Lee2,4, anuria and an elevated serum creatinine level. Her medical history showed primary 3,4 3,4 Jong-Ryeol Choi , Kyu-Sup Lee , infertility with diffuse adenomyosis. On admission, her pregnancy test was negative and 1,4 and Ihm Soo Kwak her menstrual cycle had started 1 day previously. Laboratory data were consistent with DIC, Department of Internal Medicine1, Division of and it was believed to be related to myometrial injury resulting from heavy intramyometrial Nephrology, Department of Internal Medicine2, menstrual flow. Gonadotropin is considered to play an important role in the development Division of Infectious Disease, Department of of fulminant DIC. This rare case suggests that physicians should be aware that gonadotropin Obstetrics and Gynecology3, Medical Research may provoke fulminant DIC in women with adenomyosis. Institute4, Pusan National University School of Medicine, Busan, Korea Key Words: Kidney Failure; Multiple Organ Failure; Disseminated Intravascular Coagulation; Received: 11 September 2009 Menstruation; Gonadotropins Accepted: 13 November 2009 Address for Correspondence: Dong Won Lee, M.D. -

Advances in Molecular Electronics: a Brief Review
Open Access International Journal of Women's Health and Gynecology Volume 1 Issue 1 ISSN: 2694-4081 Case Report Idiopathic Thrombocytopenic Purpura with Inaugural Manifestation Being a Heavy Menstrual Bleeding: A Case Report Nkwabong Ea*, Tayou Cb, Borassi Sc and Banmi LNd aAssociate Professor, Department of Obstetrics and Gynecology, Faculty of Medicine and Biomedical Sciences and University Teaching Hospital, Yaoundé, Cameroon bAssociate Professor, Hematologist, Department of Hematology, Faculty of Medicine and Biomedical Sciences and University Teaching Hospital, Yaoundé, Cameroon cResident, Faculty of Medicine and Biomedical Sciences, Yaoundé, Cameroon d Department of Obstetrics and Gynecology, Gyneco-Obstetric and Pediatric Hospital, Yaoundé, Cameroon Article Info Abstract Article History: Background: Abnormal uterine bleeding (AUB) is now classified using PALM-COEIN acronym. Low Received: 08 April, 2019 3 Accepted: 24 April, 2019 platelet count is a rare cause of coagulopathy in cases of AUB. With a platelet count above 30,000/mm , Published: 30 April, 2019 idiopathic thrombocytopenic purpura (ITP) is usually asymptomatic. The author hereby presents a case 3 of heavy menstrual bleeding (HMB) due to ITP with a thrombocytopenia of 37,000/mm and even * Corresponding author: Nkwabong 49,000/mm3. E, Associate Professor, Department of Obstetrics and Gynecology, Faculty of Case presentation: A 43-year-old black African woman with anaemia and thrombocytopenia was Medicine and Biomedical Sciences and referred to us for persistent menstrual bleeding despite transfusion of fresh blood. Her past history University Teaching Hospital, revealed a chronic gastric pain and no family history of coagulopathy. Seventeen days after the beginning Yaoundé, Cameroon; Tel: +(237) of HMB, she developed petechiae and bruises on the lower lip of the mouth and the left arm respectively. -
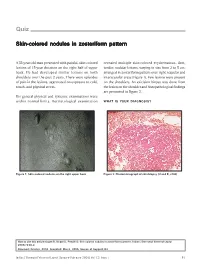
Skin-Colored Nodules in Zosteriform Pattern
Quiz Skin-colored nodules in zosteriform pattern A 33-year-old man presented with painful, skin-colored revealed multiple skin-colored erythematous, firm, lesions of 15-year duration on the right half of upper tender, nodular lesions, varying in size from 2 to 5 cm, back. He had developed similar lesions on both arranged in zosteriform pattern over right scapular and shoulders over the past 2 years. There were episodes interscapular areas (Figure 1). Few lesions were present of pain in the lesions, aggravated on exposure to cold, on the shoulders. An exicision biopsy was done from touch, and physical stress. the lesion on the shoulder and histopathological findings are presented in Figure 2. His general physical and systemic examinations were within normal limits. Dermatological examination WHAT IS YOUR DIAGNOSIS? Figure 1: Skin-colored nodules on the right upper back Figure 2: Photomicrograph of skin biopsy (H and E, x100) How to cite this article: Gupta R, Singal A, Pandhi D. Skin colored nodules in zosteriforms pattern. Indian J Dermatol Venereol Leprol 2005;72:81-2. Received: October, 2004. Accepted: March, 2005. Source of Support: Nil. Indian J Dermatol Venereol Leprol|January-February 2006|Vol 72|Issue 1 81 Quiz Diagnosis: Type-2 segmental leiomyoma cutis via sympathetic nervous system. Muscle contraction ensues, triggered by the influx of calcium ions. Hence, Histopathological findings nifedipine, a calcium channel blocker has a role in Biopsy showed a tumor composed of irregularly relieving pain associated with cutaneous leiomyoma. arranged smooth-muscle fibers, with elongated nuclei and rounded ends, interlaced with variable amounts of Several conditions are associated with piloleiomyoma,[4] collagen in the reticular dermis.