Splicing Repression Is a Major Function of Tdp-43 in Motor Neurons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
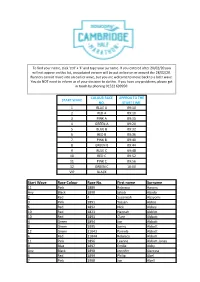
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

Ever 2018 Eupo 2018
European Association for Vision and Eye Research European University Professors of Ophthalmology EVER 2018 Annual Congress October 4-6, 2018 EUPO 2018 Course on Retina, Intraocular Inflammation & Uveitis October 3-4, 2018 Programme book Nice, France www.ever.be www.eupo.eu European Association for Vision and Eye Research EVER 20October 17-1919 in Nice, France www.ever.be 1 Table of contents Word from the president ....................................................................................................................................2 About EVER ..........................................................................................................................................................3 EVER Membership ...............................................................................................................................................4 Speakers’ affiliation to scientific sections .........................................................................................................5 Composition of the board 2018 .........................................................................................................................8 Venue ................................................................................................................................................................... 10 Congress information ....................................................................................................................................... 11 Programme information ....................................................................................................................................15 -

Short-Term Rapamycin Persistently Improves Cardiac Function After Cessation of Treatment in Aged Male and Female Mice
Short-term rapamycin persistently improves cardiac function after cessation of treatment in aged male and female mice. Ellen Quarles A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2017 Reading Committee: Peter Rabinovitch, Chair Michael MacCoss David Marcinek Program Authorized to Offer Degree: Pathology © Copyright 2017 Ellen Quarles University of Washington Abstract Short-term rapamycin persistently improves cardiac function after cessation of treatment in aged male and female mice. Ellen Quarles Chair of the Supervisory Committee: Peter Rabinovitch, Professor and Vice Chair of Research Department of Pathology Cardiac aging is an intrinsic process that results in impaired cardiac function and dysregulation of cellular and molecular quality control mechanisms. These effects are evident in the decline of diastolic function, increase in left ventricular hypertrophy, metabolic substrate shifts, and alterations to the cardiac proteome. This thesis covers the quality control mechanisms that are associated with cardiac aging, results from an anti-aging intervention in aged mice, and a review of mitochondrial dysfunction in the heart. Chapter one is a review of the quality control mechanisms in aging myocardium. Chapter two consists of the results of several mouse experiments that compare the cardiac function, proteomes, and metabolomes of aged and young controls, along with rapamycin treated aged mice. The novelty of this study comes from the inclusion of a group of animals treated only transiently with the drug, then followed for eight weeks post-drug-removal. This persistence cohort may hold clues to deriving long-lasting benefits of rapamycin with only transient treatment. -

Kwayedza N to R
KWAYEDZA N TO R INITIA SURNAME L FIRST NAME SERIAL NUMBER NAGAR R RAMESH 104084666 NAGAR S SASIKANT 149344892 NAGAR D DHANSUK 278926066 NAGO B BENSON 101437856 NAGO F FURENGI 118481701 NAGO JP JOHN PESANAI 130561852 NAGO JJ JOB JOBE 260820489 NAGO J J 168341413 NAGOMA A ALBERT 161534153 NAIDOO M MARIA 110816624 NAIDOO DA DAVID ANTONY 140600693 NAIDOO VPR V P R 181904449 NAIDOO H HANIFA 285061524 NAIK RM RAJENDRARAI MANIBHAI 229175250 NAKA E EMERENCIA 295063968 NAKAZA AM ANISTO MARUVA 282906142 NAKWERE R RADIAS 192364649 NAMAZONJO FR FUNGAI RINOS 295154165 NAME C CLARIS 248223925 NAMUSI CW CORNELIUS WONDER 235286619 NAMUSI M MIRRIAM 255122978 NAMVULA J JEFFREY 229256229 NANDA A A 137562144 NANGARA O OBERT 267377744 NANGI M MARIYA 100220924 NANILAKU FB FREDRICK BRIGHT 216429807 NAPE N NOMORE 160447635 NAPIER RB R B 288735260 NAPOSE SD SD 249790556 NARAN V VANMARI 259529626 NARE K KHOMOTSO 120372348 NARE S SAVIOUS 132437610 NARE M MARIGOLD 261844999 NARH VH VINCENT HAYES 149324371 NASASARA RB RUTENDO BERNADINE 162197099 NASASARA E EMELLIA 109555707 NASH MJ MJ 150767426 NASHO J JAMES 125149859 NASHO KM KUNDAYI 231606080 NATHOO KJ KUSUM JACKISON 112620114 NATHOO J JAYSHREE 142868740 NATISIYO S SEREVESTON 189626028 NATO T TSIKUDZAKUENDA 253899386 NATO J JUNIOR 299846770 NAUDE D DENZIL 124564408 NAUDE SS SUSSANA SOPHIA 128542806 NAUDE E EDUAN 168389523 NAUDE TJH THEUNNIS JOHANN 239938831 NAUDE DF DUDLEY FREDERIC 172467357 NAZARE RB REGGIES BATSIRAI 243486252 NAZOMBE C CHARLES 285786978 NCELE W WILSON 112568790 NCHENGA AK ANDERSON KAROTA 144981024 -

Given Name Alternatives for Irish Research
Given Name Alternatives for Irish Research Name Abreviations Nicknames Synonyms Irish Latin Abigail Abig Ab, Abbie, Abby, Aby, Bina, Debbie, Gail, Abina, Deborah, Gobinet, Dora Abaigeal, Abaigh, Abigeal, Gobnata Gubbie, Gubby, Libby, Nabby, Webbie Gobnait Abraham Ab, Abm, Abr, Abe, Abby, Bram Abram Abraham Abrahame Abra, Abrm Adam Ad, Ade, Edie Adhamh Adamus Agnes Agn Aggie, Aggy, Ann, Annot, Assie, Inez, Nancy, Annais, Anneyce, Annis, Annys, Aigneis, Mor, Oonagh, Agna, Agneta, Agnetis, Agnus, Una Nanny, Nessa, Nessie, Senga, Taggett, Taggy Nancy, Una, Unity, Uny, Winifred Una Aidan Aedan, Edan, Mogue, Moses Aodh, Aodhan, Mogue Aedannus, Edanus, Maodhog Ailbhe Elli, Elly Ailbhe Aileen Allie, Eily, Ellie, Helen, Lena, Nel, Nellie, Nelly Eileen, Ellen, Eveleen, Evelyn Eibhilin, Eibhlin Helena Albert Alb, Albt A, Ab, Al, Albie, Albin, Alby, Alvy, Bert, Bertie, Bird,Elvis Ailbe, Ailbhe, Beirichtir Ailbertus, Alberti, Albertus Burt, Elbert Alberta Abertina, Albertine, Allie, Aubrey, Bert, Roberta Alberta Berta, Bertha, Bertie Alexander Aler, Alexr, Al, Ala, Alec, Ales, Alex, Alick, Allister, Andi, Alaster, Alistair, Sander Alasdair, Alastar, Alsander, Alexander Alr, Alx, Alxr Ec, Eleck, Ellick, Lex, Sandy, Xandra, Zander Alusdar, Alusdrann, Saunder Alfred Alf, Alfd Al, Alf, Alfie, Fred, Freddie, Freddy Albert, Alured, Alvery, Avery Ailfrid Alberedus, Alfredus, Aluredus Alice Alc Ailse, Aisley, Alcy, Alica, Alley, Allie, Allison, Alicia, Alyssa, Eileen, Ellen Ailis, Ailise, Aislinn, Alis, Alechea, Alecia, Alesia, Aleysia, Alicia, Alitia Ally, -

Hearst Corporation Los Angeles Examiner Photographs, Negatives and Clippings--Portrait Files (N-Z) 7000.1C
http://oac.cdlib.org/findaid/ark:/13030/c8w37tqm No online items Hearst Corporation Los Angeles Examiner photographs, negatives and clippings--portrait files (N-Z) 7000.1c Finding aid prepared by Rebecca Hirsch. Data entry done by Nikita Lamba, Siria Meza, Stephen Siegel, Brian Whitaker, Vivian Yan and Lindsey Zea The processing of this collection and the creation of this finding aid was funded by the generous support of the Council on Library and Information Resources. USC Libraries Special Collections Doheny Memorial Library 206 3550 Trousdale Parkway Los Angeles, California, 90089-0189 213-740-5900 [email protected] 2012 April 7000.1c 1 Title: Hearst Corporation Los Angeles Examiner photographs, negatives and clippings--portrait files (N-Z) Collection number: 7000.1c Contributing Institution: USC Libraries Special Collections Language of Material: English Physical Description: 833.75 linear ft.1997 boxes Date (bulk): Bulk, 1930-1959 Date (inclusive): 1903-1961 Abstract: This finding aid is for letters N-Z of portrait files of the Los Angeles Examiner photograph morgue. The finding aid for letters A-F is available at http://www.usc.edu/libraries/finding_aids/records/finding_aid.php?fa=7000.1a . The finding aid for letters G-M is available at http://www.usc.edu/libraries/finding_aids/records/finding_aid.php?fa=7000.1b . creator: Hearst Corporation. Arrangement The photographic morgue of the Hearst newspaper the Los Angeles Examiner consists of the photographic print and negative files maintained by the newspaper from its inception in 1903 until its closing in 1962. It contains approximately 1.4 million prints and negatives. The collection is divided into multiple parts: 7000.1--Portrait files; 7000.2--Subject files; 7000.3--Oversize prints; 7000.4--Negatives. -

BASEBALL 2007 Roster 5 2007 Preview 6 Schedule 2007 Seawolves 7-14 DATE OPPONENT LOCATION TIME 2006 Review Fri-Feb
Table of Contents 2007 Information 2007 Seawolves Schedule 1 Head Coach John Goelz 2 Assistant Coaches 3-4 BASEBALL 2007 Roster 5 2007 Preview 6 Schedule 2007 Seawolves 7-14 DATE OPPONENT LOCATION TIME 2006 Review Fri-Feb. 2 CAL STATE MONTEREY BAY ROHNERT PARK 2:00 pm 2006 Highlights 16 Sat-Feb. 3 CAL STATE MONTEREY BAY (dh) ROHNERT PARK 11:00 am 2006 Results 16 Tue-Feb. 6 SAN FRANCISCO STATE ROHNERT PARK 2:00 pm 2006 Statistics 17-19 Thu-Feb. 8 WESTERN OREGON ROHNERT PARK 2:00 pm 2006 Honors 19 Sat-Feb. 10 Western Oregon San Francisco, Calif 2:00 pm 2006 CCAA Standings 20 Fri-Feb. 16 Grand Canyon Phoenix, Ariz 5:00 pm 2006 All-Conference Honors 20 2006 CCAA Leaders 21 Sat-Feb. 17 Grand Canyon (dh) Phoenix, Ariz 12:00 pm Sun-Feb. 18 Grand Canyon Phoenix, Ariz 11:00 am SSU Baseball History Sat-Feb. 24 CAL STATE STANISLAUS* (dh) ROHNERT PARK 11:00 am Yearly Team Honors 22 Sun-Feb. 25 CAL STATE STANISLAUS* (dh) ROHNERT PARK 11:00 am All-Time Awards 22-23 Fri-Mar. 2 UC SAN DIEGO* ROHNERT PARK 2:00 pm All-Americans 24 Sat-Mar. 3 UC SAN DIEGO* (dh) ROHNERT PARK 11:00 am All-Time SSU Baseball Team 25 Sun-Mar. 4 UC SAN DIEGO* ROHNERT PARK 11:00 am Yearly Leaders 26-27 Fri-Mar. 9 Cal State Dominguez Hills* Carson, Calif 2:00 pm Records 28-30 Sat-Mar. 10 Cal State Dominguez Hills* (dh) Carson, Calif 11:00 am Yearly Team Statistics 30 Sun-Mar. -

2009 National JX Qualifiers
JX Qualifiers 2008/09 - Search by Surname Last Name FirstName JX Standard Age Club State Abagi Mitchell Gold 13 Barker Aquatic NSW Abagi Curtis Silver 10 Barker Aquatic NSW Abbey Luke Silver 10 Mount Annan NSW Abbey Josh Green 12 H2O VIC Abbott Claudia Gold Star 10 AB Thunderbolts QLD Abbott Samantha Gold 13 Revesby Workers NSW Abbott Sarah Green 10 Cruiz NSW Abbott Dean Green 13 Marlin Coast QLD Abbott Jordan Green 9 AB Thunderbolts QLD Abdalian Celine Silver 11 MLC VIC Abdi Aydin Silver 11 North West Aquatic VIC Abdilla Chloe Silver 11 Epping VIC Abel Rebekah Bronze 9 Raymond Terrace NSW Abela Jasmine Silver 13 Southport Olympic QLD Abela Jaya Green 9 Werribee VIC Abels Nicolas Green 11 CA Tritons VIC Abetz Jeremy Green 13 The Hobart Aquatic Club TAS Abeya Harrison Gold Star 9 East Brisbane QLD Abraham Dora Gold 13 Elizabeth Aquatic SA Abraham Lauren Green 12 Canberra NSW Abrahams Georgia Bronze 10 Hunter NSW Accardi Hannah Silver 13 Warragul VIC Achilles Dominique Silver 11 Fairholme QLD Acitelli Anthony Green 13 Macquarie Fields NSW Ackermann Ellise Silver 13 Warwick QLD Adam Cody Gold Star 9 Carlile NSW Adam Tyler Bronze 11 Carlile NSW Adamovic Emily Green 13 Southside Aquatics QLD Adamovic Isobel Green 11 Southside Aquatics QLD Adams Olivia Gold Star 9 Ravenswood NSW Adams Joanna Gold Star 11 Ravenswood NSW Adams Jessica Gold Star 10 Yarra Plenty VIC Adams Rachel Gold 10 SOPAC Swim Club NSW Adams Katherine Gold 13 Ravenswood NSW Adams Ella Gold 13 Launceston Aquatic Club TAS Adams James Gold 13 Kings Mornington VIC Adams Ryan Gold -

A Case of Long QT Syndrome
CASE REPORT A case of long QT syndrome: challenges on a bumpy road Peter Magnusson1,2 & Per-Erik Gustafsson2 1Cardiology Research Unit, Department of Medicine, Karolinska Institutet, Stockholm SE-171 76, Sweden 2Centre for Research and Development, Uppsala University/Region Gavleborg,€ Gavle€ SE-801 87, Sweden Correspondence Key Clinical Message Peter Magnusson, Cardiology Research Unit, Department of Medicine, Karolinska Beta-agonist treatment during pregnancy may unmask the diagnosis of long QT Institutet, Karolinska University Hospital/ syndrome. The QT prolongation can result in functional AV block. A history of Solna, SE-171 76 Stockholm, Sweden. Tel: seizure and/or sudden death in a family member should raise suspicion of ven- +46(0)705 089407; Fax: +46(0)26 154255; tricular tachycardia. More than one mutation may coexist. Refusal of beta- E-mail: [email protected] blocker therapy complicates risk stratification. Funding Information Keywords No sources of funding were declared for this Genetic, implantable cardioverter–defibrillator, long QT syndrome, pregnancy, study. premature ventricular complex, risk stratification, sudden cardiac death. Received: 16 December 2016; Revised: 29 March 2017; Accepted: 4 April 2017 Clinical Case Reports 2017; 5(6): 954–960 doi: 10.1002/ccr3.985 Introduction experienced palpitations, and her ECG was abnormal, revealing PVCs, atrioventricular (AV) 2:1 block and QT Long QT syndrome (LQTS) is linked to mutations in the prolongation (520 msec) in precordial lead V5 during ion channels, which can lead to disturbances in ventricu- sinus rhythm at 90 beats per minute (Fig. 1) and rhythm lar repolarization [1]. This condition puts patients at risk strip while walking (Fig. -

SMM List.Xlsx
Profession Description Rank Code License Number Last Name First Name License Status Code License Status Description Expire Date Specialty Board Specialty Certification Medical Doctor ME 6540 GRAHAM ANGUS 20 CLEAR 01/31/2020 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 7687 PLATT MARVIN 20 CLEAR 01/31/2020 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 8046 WEISE EDMUND 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 8466 ROOT ALLEN 20 CLEAR 01/31/2020 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 9107 BECKMEYER WILLIAM 20 CLEAR 01/31/2020 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 9585 SCHIEBLER GEROLD 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 9756 GESSNER IRA 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 9890 QUILLIAN WARREN 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 10046 FISCH GILBERT 20 CLEAR 01/31/2020 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 10538 PITISCI DONALD 20 CLEAR 01/31/2020 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 10821 STEINER MICHAEL 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 10969 MARCADIS ISAAC 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 11002 RICHARDSON BOBBY 20 CLEAR 01/31/2021 AMERICAN BOARD OF PEDIATRICS PD ‐ PEDIATRICS Medical Doctor ME 11621 CIMINO DAVID 20 CLEAR 01/31/2021 -

South Australian Government Boards and Committees Information As at 30 June 2020
OFFICIAL South Australian Government Boards and Committees Information As at 30 June 2020 OFFICIAL OFFICIAL South Australian Government Boards and Committees As at 30 June 2020 Introduction This is the 24th annual report to Parliament of consolidated South Australian Government board and committee information1. The report sets out the membership and remuneration arrangements of 196 part-time government boards and committees as at 30 June 2020 in order of ministerial portfolio. The information has been sourced from the Boards and Committees Information System (BCIS), a database administered by the Department of the Premier and Cabinet, following extensive consultation with all ministerial offices and stakeholder agencies. Definition of boards and committees in the report The boards and committees included in this report are those which are: • established by or under an Act of Parliament of South Australia (generally excluding the Local Government Act 1999) and have a majority of members appointed by either a minister or the Governor; or • established by a minister or legal instrument such as a constitution or charter, have a majority of members appointed by a minister, and have at least one member in receipt of remuneration. The report should not be considered to be a complete listing of all government boards and committees. 1 Note: The 2019 report, which was the 23rd such report, was incorrectly published as the 22nd report. This error dates to the 2014 annual report, which was incorrectly published as the 17th report, resulting in all subsequent reports being incorrectly labelled. This error has been corrected in the copies of the report available on the DPC website Page 2 of 9 OFFICIAL OFFICIAL Highlights Number of boards and committees 196 boards and committees are identified in the 2020 report. -

In Re Network Associates, Inc. Securities Litigation 00-CV-4849
MC66N Rejected or Ineligible Claimants Page 1 of 251 MC66N138 NETWORK ASSOCIATES, INC. II SECUR REPS 13-Jun-05 11:50 AM Reason Deemed Claim Number Name City State Ineligible 10559 WATKINS, JOYCE E ATLANTA GA FATAL LINE 12498 1199 HEALTH CARE EMP CHICAGO IL NO LOSS 7784 16105 PIMCO IARORCIH NEW YORK NY NO LOSS 2259 20 UIT FUNDS NEWYORK NY INTRINSICALLY INELIGIBLE 2109830 33 WEST CLINTON AVEN TOMS RIVER NJ NO LOSS 9826 3M ERIP TRUST PITTSBURGH PA FATAL LINE 8466 4 Z'S INVESTMENTS PA WATERFORD MI NO LOSS 1080 45 INDIANA HOSPITOL PHILA PA NO LOSS 8779 777-H F II 1990 STLM DETROIT MI NO LOSS 2093721 800 PRE-EMPTION ROAD GENEVA NY NO LOSS 2247 91325 CANADA INC NO LOSS 8375 A DIAMOND FAMILY TRU LOS ANGELES CA NO LOSS 2065510 A F & N CIRAMELLA RE DAYTON OH NO LOSS 2029983 A R W SUPPORT TRUST TAYLORVILLE IL INTRINSICALLY INELIGIBLE 12103 AAA MI RESTRUCTURING CHICAGO IL NO LOSS 2093159 AAOF ENDOWMENT FUND SAINT LOUIS MO NO LOSS 2095827 AARON, BRIAN & CAROL BETHESDA MD FATAL LINE 2091134 ABAD, MARIO & NAVATA WEST ORANGE NJ NO LOSS 2027060 ABBAMONTE, MICHAEL & SOLON OH INTRINSICALLY INELIGIBLE 2101711 ABBETT, DEANNA L SAN JOSE CA FATAL LINE 2055264 ABDELQADER, MAHMUD S COLLINSVILLE IL NO LOSS 2030513 ABEGGLEN, CHERYL ROS CHADRON NE INTRINSICALLY INELIGIBLE 4336 ABEL, DAVID MELVILLE NY NO LOSS 2015464 ABEL, KEVIN KEW GARDENS NY NO LOSS 2020708 ABEL, STEPHEN ONEIDA NY NO LOSS 8542 ABEUTT, BETWORT MERRICK NY NO LOSS 2127049 ABLEY, PETER & VIBEK NO LOSS 6756 ABN AMRO N QUINCY MA INTRINSICALLY INELIGIBLE 4204 ABN AMRO SMALL CAP F CHICAGO IL NO LOSS 1214