The Cone Dysfunction Syndromes Jonathan Aboshiha,1,2 Adam M Dubis,1,2 Joseph Carroll,3 Alison J Hardcastle,1,2 Michel Michaelides1,2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Blue Cone Monochromacy: Visual Function and Efficacy Outcome Measures for Clinical Trials
RESEARCH ARTICLE Blue Cone Monochromacy: Visual Function and Efficacy Outcome Measures for Clinical Trials Xunda Luo1☯‡, Artur V. Cideciyan1☯‡*, Alessandro Iannaccone2, Alejandro J. Roman1, Lauren C. Ditta2, Barbara J. Jennings2, Svetlana A. Yatsenko3, Rebecca Sheplock1, Alexander Sumaroka1, Malgorzata Swider1, Sharon B. Schwartz1, Bernd Wissinger4, Susanne Kohl4, Samuel G. Jacobson1* 1 Scheie Eye Institute, Department of Ophthalmology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, United States of America, 2 Hamilton Eye Institute, Department of Ophthalmology, University of Tennessee Health Science Center, Memphis, Tennessee, United States of America, 3 Pittsburgh Cytogenetics Laboratory, Center for Medical Genetics and Genomics, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, United States of America, 4 Molecular Genetics Laboratory, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tuebingen, Tuebingen, Germany ☯ These authors contributed equally to this work. ‡ OPEN ACCESS These authors are joint first authors on this work. * [email protected] (SGJ); [email protected] (AVC) Citation: Luo X, Cideciyan AV, Iannaccone A, Roman AJ, Ditta LC, Jennings BJ, et al. (2015) Blue Cone Monochromacy: Visual Function and Efficacy Abstract Outcome Measures for Clinical Trials. PLoS ONE 10(4): e0125700. doi:10.1371/journal.pone.0125700 Academic Editor: Dror Sharon, Hadassah-Hebrew University Medical Center, ISRAEL Background Blue Cone Monochromacy (BCM) is an X-linked retinopathy caused by mutations in the Received: December 29, 2014 OPN1LW / OPN1MW gene cluster, encoding long (L)- and middle (M)-wavelength sensitive Accepted: March 21, 2015 cone opsins. Recent evidence shows sufficient structural integrity of cone photoreceptors in Published: April 24, 2015 BCM to warrant consideration of a gene therapy approach to the disease. -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Updates on Myopia
Updates on Myopia A Clinical Perspective Marcus Ang Tien Y. Wong Editors Updates on Myopia Marcus Ang • Tien Y. Wong Editors Updates on Myopia A Clinical Perspective Editors Marcus Ang Tien Y. Wong Singapore National Eye Center Singapore National Eye Center Duke-NUS Medical School Duke-NUS Medical School National University of Singapore National University of Singapore Singapore Singapore This book is an open access publication. ISBN 978-981-13-8490-5 ISBN 978-981-13-8491-2 (eBook) https://doi.org/10.1007/978-981-13-8491-2 © The Editor(s) (if applicable) and The Author(s) 2020, corrected publication 2020 Open Access This book is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this book are included in the book's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the book's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specifc statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -

Colour Vision Deficiency
Eye (2010) 24, 747–755 & 2010 Macmillan Publishers Limited All rights reserved 0950-222X/10 $32.00 www.nature.com/eye Colour vision MP Simunovic REVIEW deficiency Abstract effective "treatment" of colour vision deficiency: whilst it has been suggested that tinted lenses Colour vision deficiency is one of the could offer a means of enabling those with commonest disorders of vision and can be colour vision deficiency to make spectral divided into congenital and acquired forms. discriminations that would normally elude Congenital colour vision deficiency affects as them, clinical trials of such lenses have been many as 8% of males and 0.5% of femalesFthe largely disappointing. Recent developments in difference in prevalence reflects the fact that molecular genetics have enabled us to not only the commonest forms of congenital colour understand more completely the genetic basis of vision deficiency are inherited in an X-linked colour vision deficiency, they have opened the recessive manner. Until relatively recently, our possibility of gene therapy. The application of understanding of the pathophysiological basis gene therapy to animal models of colour vision of colour vision deficiency largely rested on deficiency has shown dramatic results; behavioural data; however, modern molecular furthermore, it has provided interesting insights genetic techniques have helped to elucidate its into the plasticity of the visual system with mechanisms. respect to extracting information about the The current management of congenital spectral composition of the visual scene. colour vision deficiency lies chiefly in appropriate counselling (including career counselling). Although visual aids may Materials and methods be of benefit to those with colour vision deficiency when performing certain tasks, the This article was prepared by performing a evidence suggests that they do not enable primary search of Pubmed for articles on wearers to obtain normal colour ‘colo(u)r vision deficiency’ and ‘colo(u)r discrimination. -

Clinical and Genetic Investigation of a Large Tunisian Family with Complete Achromatopsia: Identification of a New Nonsense Mutation in GNAT2 Gene
Journal of Human Genetics (2011) 56, 22–28 & 2011 The Japan Society of Human Genetics All rights reserved 1434-5161/11 $32.00 www.nature.com/jhg ORIGINAL ARTICLE Clinical and genetic investigation of a large Tunisian family with complete achromatopsia: identification of a new nonsense mutation in GNAT2 gene Farah Ouechtati1,2,7, Ahlem Merdassi2,7, Yosra Bouyacoub1,2, Leila Largueche2, Kaouther Derouiche2, Houyem Ouragini1, Sonia Nouira1, Leila Tiab3,4, Karim Baklouti2, Ahmed Rebai5, Daniel F Schorderet3,4,6, Francis L Munier3,4,6, Leonidas Zografos4,6, Sonia Abdelhak1 and Leila El Matri2 Complete achromatopsia is a rare autosomal recessive disease associated with CNGA3, CNGB3, GNAT2 and PDE6C mutations. This retinal disorder is characterized by complete loss of color discrimination due to the absence or alteration of the cones function. The purpose of the present study was the clinical and the genetic characterization of achromatopsia in a large consanguineous Tunisian family. Ophthalmic evaluation included a full clinical examination, color vision testing and electroretinography. Linkage analysis using microsatellite markers flanking CNGA3, CNGB3, GNAT2 and PDE6C genes was performed. Mutations were screened by direct sequencing. A total of 12 individuals were diagnosed with congenital complete achromatopsia. They are members of six nuclear consanguineous families belonging to the same large consanguineous family. Linkage analysis revealed linkage to GNAT2. Mutational screening of GNAT2 revealed three intronic variations c.119À69G4C, c.161+66A4T and c.875À31G4C that co-segregated with a novel mutation p.R313X. An identical GNAT2 haplotype segregating with this mutation was identified, indicating a founder mutation. All patients were homozygous for the p.R313X mutation. -

Structure of Cone Photoreceptors
Progress in Retinal and Eye Research 28 (2009) 289–302 Contents lists available at ScienceDirect Progress in Retinal and Eye Research journal homepage: www.elsevier.com/locate/prer Structure of cone photoreceptors Debarshi Mustafi a, Andreas H. Engel a,b, Krzysztof Palczewski a,* a Department of Pharmacology, Case Western Reserve University, Cleveland, OH 44106-4965, USA b Center for Cellular Imaging and Nanoanalytics, M.E. Mu¨ller Institute, Biozentrum, WRO-1058, Mattenstrasse 26, CH 4058 Basel, Switzerland abstract Keywords: Although outnumbered more than 20:1 by rod photoreceptors, cone cells in the human retina mediate Cone photoreceptors daylight vision and are critical for visual acuity and color discrimination. A variety of human diseases are Rod photoreceptors characterized by a progressive loss of cone photoreceptors but the low abundance of cones and the Retinoids absence of a macula in non-primate mammalian retinas have made it difficult to investigate cones Retinoid cycle directly. Conventional rodents (laboratory mice and rats) are nocturnal rod-dominated species with few Chromophore Opsins cones in the retina, and studying other animals with cone-rich retinas presents various logistic and Retina technical difficulties. Originating in the early 1900s, past research has begun to provide insights into cone Vision ultrastructure but has yet to afford an overall perspective of cone cell organization. This review Rhodopsin summarizes our past progress and focuses on the recent introduction of special mammalian models Cone pigments (transgenic mice and diurnal rats rich in cones) that together with new investigative techniques such as Enhanced S-cone syndrome atomic force microscopy and cryo-electron tomography promise to reveal a more unified concept of cone Retinitis pigmentosa photoreceptor organization and its role in retinal diseases. -

The Genetic Horizon Improving Clinical Sensitivity in Difficult-To-Sequence Genes for Rare Hereditary Disorders
White paper The Genetic Horizon Improving clinical sensitivity in difficult-to-sequence genes for rare hereditary disorders Meeting clinical needs using custom solutions From Bench to Bedside: The Genetic Horizon Ongoing challenges The long-awaited promise of tailored treatments for individual patients based on their genetic Clinically relevant but highly homologous and · Masked regions in the reference data sets contain makeup is beginning to materialize. Next generation sequencing, along with artificial intelligence, repetitive regions within important genes are an uncertain or ambiguous variant calls that are the source ongoing challenge to analyze: of false positives, false negatives and other genotype have facilitated the rapid, accurate analysis and interpretation of genomic data for disease causing calling errors (e.g., calling homozygous variant at a variants which may be treatable through emerging gene therapy technology. · Certain genomic regions are difficult to analyze due to heterozygous site)1. complex sequence variations therein The number of clinical trials and drug development involves delivering a functional copy of the SMN1 gene in a Blueprint Genetics remains committed to resolving pipelines for rare disease are quickly growing and, one-time intravenous infusion (www.avexis.com). · Not only are there variants in these regions but there difficult-to-sequence regions that are hard to validate, encouragingly, the first approved treatments have been are types of variants that we cannot detect in the clinical interpret and confirm, by developing custom solutions. In successful. As the field of precision medicine is still in its The power of gene therapies is in the specificity of the diagnostic laboratory with current technologies (Table 1) this paper, we share our strategies and what is next in our infancy, increased awareness about the clinical utility of treatments: understanding the genetic mechanism of R&D pipeline. -

Intermixing the OPN1LW and OPN1MW Genes Disrupts the Exonic Splicing Code Causing an Array of Vision Disorders
G C A T T A C G G C A T genes Review Intermixing the OPN1LW and OPN1MW Genes Disrupts the Exonic Splicing Code Causing an Array of Vision Disorders Maureen Neitz * and Jay Neitz Department of Ophthalmology and Vision Science Center, University of Washington, Seattle, WA 98109, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-206-543-7998 Abstract: Light absorption by photopigment molecules expressed in the photoreceptors in the retina is the first step in seeing. Two types of photoreceptors in the human retina are responsible for image formation: rods, and cones. Except at very low light levels when rods are active, all vision is based on cones. Cones mediate high acuity vision and color vision. Furthermore, they are critically important in the visual feedback mechanism that regulates refractive development of the eye during childhood. The human retina contains a mosaic of three cone types, short-wavelength (S), long-wavelength (L), and middle-wavelength (M) sensitive; however, the vast majority (~94%) are L and M cones. The OPN1LW and OPN1MW genes, located on the X-chromosome at Xq28, encode the protein component of the light-sensitive photopigments expressed in the L and M cones. Diverse haplotypes of exon 3 of the OPN1LW and OPN1MW genes arose thru unequal recombination mechanisms that have intermixed the genes. A subset of the haplotypes causes exon 3- skipping during pre-messenger RNA splicing and are associated with vision disorders. Here, we review the mechanism by which splicing defects in these genes cause vision disorders. Citation: Neitz, M.; Neitz, J. -

Progressive Cone and Cone-Rod Dystrophies
Br J Ophthalmol: first published as 10.1136/bjophthalmol-2018-313278 on 24 January 2019. Downloaded from Review Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy Jasdeep S Gill,1 Michalis Georgiou,1,2 Angelos Kalitzeos,1,2 Anthony T Moore,1,3 Michel Michaelides1,2 ► Additional material is ABSTRact proteins involved in photoreceptor structure, or the published online only. To view Progressive cone and cone-rod dystrophies are a clinically phototransduction cascade. please visit the journal online (http:// dx. doi. org/ 10. 1136/ and genetically heterogeneous group of inherited bjophthalmol- 2018- 313278). retinal diseases characterised by cone photoreceptor PHOTORECEPTION AND THE degeneration, which may be followed by subsequent 1 PHOTOTRANSDUCTION CASCADE UCL Institute of rod photoreceptor loss. These disorders typically present Rod photoreceptors contain rhodopsin phot- Ophthalmology, University with progressive loss of central vision, colour vision College London, London, UK opigment, whereas cone photoreceptors contain 2Moorfields Eye Hospital NHS disturbance and photophobia. Considerable progress one of three types of opsin: S-cone, M-cone or Foundation Trust, London, UK has been made in elucidating the molecular genetics L-cone opsin. Disease-causing sequence variants 3 Ophthalmology Department, and genotype–phenotype correlations associated with in the genes encoding the latter two cone opsins University of California San these dystrophies, with mutations in at least 30 genes -
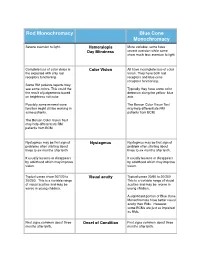
Comparison RM Versus BCM 2
Rod Monochromacy Blue Cone Monochromacy Severe aversion to light. Hemeralopia More variable- some have Day Blindness severe aversion while some show much less aversion to light. Complete loss of color vision is Color Vision All have incomplete loss of color the expected with only rod vision. They have both rod receptors functioning. receptors and blue cone receptors functioning. Some RM patients reports they see some colors. This could the Typically they have some color the result of judgements based detection along the yellow- blue on brightness not color. axis. Possibly some minimal cone The Berson Color Vision Test function might still be working in may help differentiate RM some patients. patients from BCM. The Berson Color Vision Test may help differentiate RM patients from BCM. Nystagmus may be first sign of Nystagmus Nystagmus may be first sign of problems often starting about problem often starting about three to six months after birth. three to six months after birth. It usually lessens or disappears It usually lessens or disappears by adulthood which may improve by adulthood which may improve vision. vision. Typical cases show 20/120 to Visual acuity Typical cases 20/60 to 20/200 20/200. This is a variable range This is a variable range of visual of visual acuities and may be acuities and may be worse in worse in young children. young children. A significant portion of Blue Cone Monochromats have better visual acuity than RMs. However, some BCMs are just as impaired as RMs. First signs common about three Onset of Condition First signs common about three months after birth. -
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

The Genetics of Myopia 5 Milly S
The Genetics of Myopia 5 Milly S. Tedja, Annechien E. G. Haarman, Magda A. Meester-Smoor, Virginie J. M. Verhoeven, Caroline C. W. Klaver, and Stuart MacGregor Key Points • While the recent global rise of myopia prevalence is primarily attributable to environmental changes, within populations inherited factors play a large role in explaining why some individuals are affected by myopia while oth- ers are not. • Early efforts to identify the specific genes underlying the heritability of refractive error used linkage and candidate gene designs to identify up to 50 loci and genes, although most remain unconfirmed. M. S. Tedja · A. E. G. Haarman · M. A. Meester-Smoor Department of Ophthalmology, Erasmus Medical Center, Rotterdam, The Netherlands Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands V. J. M. Verhoeven Department of Ophthalmology, Erasmus Medical Center, Rotterdam, The Netherlands Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands Department of Clinical Genetics, Erasmus Medical Center, Rotterdam, The Netherlands C. C. W. Klaver Department of Ophthalmology, Erasmus Medical Center, Rotterdam, The Netherlands Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands Department of Ophthalmology, Radboud University Medical Center, Nijmegen, The Netherlands S. MacGregor (*) Statistical Genetics, QIMR Berghofer Medical Research Institute, Brisbane, QLD, Australia e-mail: [email protected] © The Author(s) 2020 95 M. Ang, T. Y. Wong (eds.), Updates on Myopia, https://doi.org/10.1007/978-981-13-8491-2_5 96 M. S. Tedja et al. • As the sample size in genome-wide association studies (GWAS) has increased, the number of implicated loci has risen steadily, with 161 vari- ants reported in the latest meta-analysis.