Chondrodysplasia-Like Dwarfism in the Miniature Horse
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

List of Horse Breeds 1 List of Horse Breeds
List of horse breeds 1 List of horse breeds This page is a list of horse and pony breeds, and also includes terms used to describe types of horse that are not breeds but are commonly mistaken for breeds. While there is no scientifically accepted definition of the term "breed,"[1] a breed is defined generally as having distinct true-breeding characteristics over a number of generations; its members may be called "purebred". In most cases, bloodlines of horse breeds are recorded with a breed registry. However, in horses, the concept is somewhat flexible, as open stud books are created for developing horse breeds that are not yet fully true-breeding. Registries also are considered the authority as to whether a given breed is listed as Light or saddle horse breeds a "horse" or a "pony". There are also a number of "color breed", sport horse, and gaited horse registries for horses with various phenotypes or other traits, which admit any animal fitting a given set of physical characteristics, even if there is little or no evidence of the trait being a true-breeding characteristic. Other recording entities or specialty organizations may recognize horses from multiple breeds, thus, for the purposes of this article, such animals are classified as a "type" rather than a "breed". The breeds and types listed here are those that already have a Wikipedia article. For a more extensive list, see the List of all horse breeds in DAD-IS. Heavy or draft horse breeds For additional information, see horse breed, horse breeding and the individual articles listed below. -

Model Equine Photo Showers Association Facebook Page
FEATURES: Volume 11 JULY-SEPT 2014 GET READY FOR THE QUARTERLY NEWSLETTER NEW SHOW SEASON EDITOR: Elizabeth Jones The Mysterious Mare! By Alexa Goudy ON PAGE 11 http://mepsa1.tripod.com/mepsa.htm MEPSA is an educational group for model horse enthusiasts, promoting the hobby of model horse mail-in photo showing. Editor note: I am typing this newsletter with one finger due to the fact I broke my arm! Annual Championship…is underway! As of this writing division A and the mini division have been judged! With any luck, the show will be completed by the end of July and the book will go to print in early August! MEPSA is an open, photo show organization. You do not need to pay any membership fees to show. No registration needed. Just send your models, return envelope and your show fee, and you will be competing with some of the best. Not quite there yet? MEPSA also has a Novice division, designed to help coach new MEPSA showers to reach the level they need to achieve to be successful in the open division! Contact Marie Phillips ([email protected]) if you have questions about entering novice shows! It’s time to prep for the new show season! Like you, I am biting my nails waiting for championship results. To distract myself I am taking new (hopefully improved) photos and labeling them for the next MEPSA season. – In support of that endeavor –this newsletter will include information on things like: where to show your halter horse, how to take winning photos, getting into the performance arena tacking up for English pleasure, etc. -

Whc Program Deadlines!
November 2017 2 Mission Statement WHC PROGRAM DEADLINES! 3 WSHCEF 4 GOP, Equine Industry Canter Toward Tax Reform - AHC It is that time of year again! Time to wind down Class yet another year and start thinking about 65 Horse2017 Trail Owners Master Can Certification Ensure Fall submitting your applications, nominations and Equine Wellness 7 Middle Inlet Horse Camp hour logs for the many WHC programs! 8 Northern Saddle Club Trail Farm Fundraiser Meets Goal Please take a moment to mark your calendars 9 HorseGrant UpdatePasture / Care Horse in FallRescue Can Pay Off All Year with the list of WHC Program deadline dates 10 JCDHA shown below. Some program submission deadlines include the WHC 11 Guest Worker Visa Reform Gains Momentum - AHC Annual Awards nomination forms, Scholarship applications, Sponsorship/ 12 Grant applications, Trail Grant applications, Ride Wisconsin Trail Ride/Drive 13 Midwest Horse Fair Classified Ads / EDCC Program Hour Logs, etc. Please don’t miss out on participating in our 14 Trail Reviews programs due to simply missing a deadline! 16 WHC Sponsorship / Grant 15 TrailProgram Reviews Information (cont.) 17 Events / “Did You Know?” WHC Equine of the Year Nomination Deadline - 12/1/17 Calendar of Upcoming 18 Annual Award Nominations Annual Awards Program Nomination Deadline - 1/10/18 Now Open Sponsorship/Grants Program Submission Deadline - 1/31/18 19 SPECIAL CUT & FOLD! Equine Owners - $1 Million Ride WI! Trail Ride/Drive Program Hour Logs Due - 1/31/18 Making a Difference for Trail Grant Submission Deadline - 2/1/18 -

Bits and Pieces: I Need to Make a Correction on My Last Newsletter
Bits and Pieces: I need to make a correction on my last newsletter. It was brought to my attention that Tracy Smith’s donation of a breeding is not to The Rookie as stated, but to his son RookiesGalantAparition, a homozygous tovero. Sorry for the mix up and thank you Tracy for this very kind donation to the youth raffle. Claudeane Killfoil sent an update on the new arrivals at her place. A dun colt half Arab/half APHA by Cotton Pickin Smoke out of an Arab mare. A grullo overo colt by Cotton Pickin Smoke out of Miss Melodys Hobby (QH), a chestnut overo colt by Wild About Who, out of Commanders Brass (QH) and one sad note that her grullo tovero filly by Cotton Pickin Smoke out of Voodoo Kiss had to be put down. So sorry Claudeane. I have a sorrel overo yearling colt by FPF Lethal Weapon out of a Cracker Jack Sonny mare here that’s for sale. He is with us for some ground work and fitting and will make someone a winner. If you have interest in this nice colt or would like more information about him give me a call, and I’ll put you in touch with the owner. He’s a nice colt, and will definitely be a winner. Mandy Brinnand emailed a sale list; A 2003 AQHA dark liver chestnut filly. Sire is A Bold Conclusion, Dam is a own daughter of Impressive NYPP N/N, $2300.00 APHA 2001 BS gelding, by A Bold Conclusion out of an own daughter of Barlink Macho Man, groundwork started, very intelligent w/great disposition, will be a rider, $1800.00 APHA 2000 sorrel BS gelding, by A Bold Conclusion out of Cute N Sizzlin, superior halter mare by Sizzlin Sonething by Sizzlin Hot. -

Programs That Extend the Useful Lives of Horses
PROGRAMS THAT EXTEND THE USEFUL LIVES OF HORSES: Supply and demand — they are the ingredients that form the foundation for near- ly all successful business models. Markets are sound and profitable when there is a healthy balance between the two. The theory holds true for the horse market as well. Often, however, owners may not be aware of the demands that exist for horses that may be “unwanted” by some, but desired by others. The purpose of this chapter is to show some of the many programs already in place by horse breed organizations and other groups, in which horses are needed by participants. From trail riding enthusiasts to horse show exhibitors, people are searching every day for horses that fit their lifestyles and interests. By understanding the activities encouraged by breed organizations, owners of some unwanted horses might find a good fit, and a good market, among people seeking horses for organized shows and rec- reational events. Following is a listing of some of the most popular programs and activities underway today and some true-life stories of unwanted horses that developed into champions. Competitive Horse Shows: Nearly all horse breed associations offer opportunities for friendly competition. Although a horse may be retired or reaching advanced years of maturity, horse shows offer outlets to help keep the horse active and involved. The registries offer several different disciplines with classes ranging from leadline to saddle seat pleasure to barrel racing. A horse owner can often find a way to keep horses involved for a long time in the variety of disciplines offered in the showing world. -
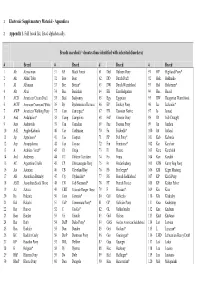
Electronic Supplementary Material - Appendices
1 Electronic Supplementary Material - Appendices 2 Appendix 1. Full breed list, listed alphabetically. Breeds searched (* denotes those identified with inherited disorders) # Breed # Breed # Breed # Breed 1 Ab Abyssinian 31 BF Black Forest 61 Dul Dülmen Pony 91 HP Highland Pony* 2 Ak Akhal Teke 32 Boe Boer 62 DD Dutch Draft 92 Hok Hokkaido 3 Al Albanian 33 Bre Breton* 63 DW Dutch Warmblood 93 Hol Holsteiner* 4 Alt Altai 34 Buc Buckskin 64 EB East Bulgarian 94 Huc Hucul 5 ACD American Cream Draft 35 Bud Budyonny 65 Egy Egyptian 95 HW Hungarian Warmblood 6 ACW American Creme and White 36 By Byelorussian Harness 66 EP Eriskay Pony 96 Ice Icelandic* 7 AWP American Walking Pony 37 Cam Camargue* 67 EN Estonian Native 97 Io Iomud 8 And Andalusian* 38 Camp Campolina 68 ExP Exmoor Pony 98 ID Irish Draught 9 Anv Andravida 39 Can Canadian 69 Fae Faeroes Pony 99 Jin Jinzhou 10 A-K Anglo-Kabarda 40 Car Carthusian 70 Fa Falabella* 100 Jut Jutland 11 Ap Appaloosa* 41 Cas Caspian 71 FP Fell Pony* 101 Kab Kabarda 12 Arp Araappaloosa 42 Cay Cayuse 72 Fin Finnhorse* 102 Kar Karabair 13 A Arabian / Arab* 43 Ch Cheju 73 Fl Fleuve 103 Kara Karabakh 14 Ard Ardennes 44 CC Chilean Corralero 74 Fo Fouta 104 Kaz Kazakh 15 AC Argentine Criollo 45 CP Chincoteague Pony 75 Fr Frederiksborg 105 KPB Kerry Bog Pony 16 Ast Asturian 46 CB Cleveland Bay 76 Fb Freiberger* 106 KM Kiger Mustang 17 AB Australian Brumby 47 Cly Clydesdale* 77 FS French Saddlebred 107 KP Kirdi Pony 18 ASH Australian Stock Horse 48 CN Cob Normand* 78 FT French Trotter 108 KF Kisber Felver 19 Az Azteca -

American Shetland Pony Club Inc 2018 AMHR Nationals September 6
American Shetland Pony Club Inc 2018 AMHR Nationals September 6-16, 2018 Built Ford Tough Livestock Complex, Expo Square Tulsa, Oklahoma Entries Close July 23, 2018 www.shetlandminiature.com 1 2018 AMHR Nationals TITLE SPONSOR 2 2018 American Miniature Horse Registry National Show Youth and Amateur Divisions Portia Sue Kalinka – Chilton, WI James McKeith – Coleman, MI Patricia Reiter, Columbus, MT Open Halter Division Rob Crater – Seattle, WA Gordon Odegard – Rockford, IL Janice Silvio – Allenton, MI Open Performance Division Patrick Derrick – Sioux Falls, SD James Knight – Garden City, MO Laurie Villalpando – Hooper, UT Futurity Judge Amateur & Jackpot Incentive Classes Richard Petty – Jacksonville, OR Mustang Arena Sandra Croote – Esperance, NY Curt Summers – Green Valley, AZ Margo Shallcross, Bulverde, TX National Show Stewards Lea Dill – Walworth, NY Randy Martin – Danvers, IL Mike Mounts – Grove City, OH 3 Official 2018 AMHR NATIONALS Photographer Photographs will be taken by Casey McBride during the sessions and will be available directly through him. Video will be provided by CPE1LiveCast (Chris Eaton). Arrangements may be made before the session or by calling. Commercial Photographers at all national shows are limited to Press Photographers Only. Press photographers (anyone taking pictures for newspapers or periodic publications) will be required to sign an agreement stating that they will abide by official AMHR Show rules and any photographs they take are to be used for editorial purposes only and are not to be sold under any circumstances. Press passes can only be obtained from the horse show office. Cameras with detachable lenses or lenses of more than two inches are prohibited. Any camera lens greater than two inches is considered professional equipment and is not permitted in the arena buildings or into the competition arenas to include seating areas. -

All Senior Questions Question Answer Source
All Senior Questions Question Answer Source Where in the digestive tract are amino acids Large intestine. HIH 710 synthesized? What unsoundness is most noticeable when Stringhalt. Ensminger, 530 backing the horse? Describe the ideal angles of the horse's front feet Front feet: 45 - 50 degrees. Beeman, 8 and hind feet. Rear feet: 50 - 60 degrees. An excessive reaction of the skin to sunlight is Photosensitivity. Veterinary Medicine, called what? 591 What term is used to describe a hoof wall angle Club foot. Curtis, 45 of 65 degrees or more? The American Paint Horse Association is devoted Paints, Quarter Horses, and Thoroughbreds. HBM strictly to stock horses and bases its registry on the blood of what 3 breeds? What do the letters CF stand for? Crude Fiber. Ensminger, 550 The common digital artery supplies blood to what Phalanges and foot. HBM parts of the horse? What is the interdental space? The gum space between the incisors and the HBM molars. Thursday, January 03, 1980 Page 1 of 95 University of Kentucky, College of Agriculture,Cooperative Extenison Service All Senior Questions Question Answer Source What color horses are more commonly prone to Gray horses. Veterinary Medicine, melanomas? 307 Most of the nutrients are found in what part of the Leaves. HBM forage plant? Excessive granulation tissue rising out of and Proud flesh. Ensminger, 527 above the edges of a wound is called what? Explain the functional difference of arteries and Arteries carry blood away from the heart to the Evans, Borton et all, veins in the horse's body. body tissues. -

Miniature Horse Show 2021
MINIATURE HORSE SHOW August 5th 6th and 7th 2021 Dept. MH Clif Hanson, Show Manager FOR SHOW INFORMATION: Sharon Hanson Phone: 406-590-2940 leave message Email: [email protected] MINIATURE HORSE MISSION STATEMENT: To provide a safe, competitive and friendly display and show atmosphere for the promotion of the Miniature Horse, and Ponies, by their breeders and owners, for the entertainment and education of Montana State Fair goers. JUDGE: Tim Parkinsen STEWARD: Glade Player Jumps/ Obstacle: Kathy Merrell Announcer: Teresa Phillips Show Manager/Ring Steward: Clif Hanson PRE-ENTRY DEADLINE: July21, 2021 2021 SCHEDULE OF EVENTS (All activities listed will be in the Pony Barn ,or show area) Tuesday, July 21st Entry forms due for all shows Wednesday, August 4th Noon-10pm Horse check-in (early arrival encouraged) 5pm Horses measured, post entries accepted 6pm Horses on display Thursday , August 5th 8am-8:30 Horses measured 9am ASPC, AMHR Show (horses on display after show) 6pm-7pm Demonstration with Morgan Merja Lanham CESMT: Massage Therapy in the Pony Barn Friday, August 6th 9am ASPC, AMHR Show (horses on display after show) 6pm-7pm Demonstration with Morgan Merja Lanham CESMT: Light Therapy in the Pony Barn Saturday, August 7th 9am ASPC, AMHR Show (horses released after classes and awards) Every effort will be made to follow the above schedule but due to unforeseen circumstances the schedule is subject to change. RULES & REGULATIONS 1. All participants at Montana State Fair events are responsible for reading the GENERAL INFORMATION and LIVESTOCK INFORMATION sections of this handbook and complying with policies listed therein. -

253 AMERICAN MINIATURE HORSE REGISTRY Driving Performance
AMERICAN MINIATURE HORSE REGISTRY Driving Performance Division Rules 5.1 Miniature Horse Driving Division – General Rules A. Guidance: The driving division was founded for the purpose of developing and furthering the art and sport of driving for pleasure. A working knowledge of and compliance with the rules are essential. B. The only person to handle the reins, under penalty of elimination, is the driver. No change of driver is per- mitted during any class. C. Dress Code: Headers, Drivers and their passengers should be dressed appropriately. Dress in the show ring is to complement the overall appearance of the unit, not take away from the appearance. 1. Hats for gentlemen are optional, except when in formal attire. 2. Formal wear should not be worn before 5 p.m. un- less stake classes are held in an afternoon perfor- mance session. 3. No strapless dresses in any driving class. Miniature Horse 4. No sandals or open toed shoes to be worn by driver or header. 5. No T-shirts or shorts. 6. No farm, individual, or animal names may be dis- played. Exception: Draft harness classes. D. Horses must be serviceably sound. E. Horses may be shown with a full mane or mane with bridle path clipped and full tail. F. Driving whips, if used, must be of suitable style, and the tip of the lash must not reach past the shoulder of the horse. Section XI - Driving Division Rules 253 G. Cross Entering: 1. Pleasure horses cannot cross-enter into Country Pleasure, Western Pleasure or Park Divisions at the same show. -

Estudio Comparativo Del Borde Dorsal Del Cuello En
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Portal de Revistas Científicas Complutenses ISSN: 1988‐2688 http://www.ucm.es/BUCM/revistasBUC/portal/modulos.php?name=Revistas2&id=RCCV&col=1 http://dx.doi.org/10.5209/rev_RCCV.2014.v8.n2.47161 Revista Complutense de Ciencias Veterinarias 2014 8(2):49-60 ESTUDIO COMPARATIVO DEL BORDE DORSAL DEL CUELLO EN EQUINOS MINIATURA COMPARATIVE STUDY OF DORSAL NECK IN EQUINE MINIATURE Abelardo Morales Briceño, Aniceto Méndez Sánchez, José Pérez Arévalo. ¹Departamento de Anatomía y Anatomía Patológica Comparadas, Edificio de Sanidad Animal, Campus de Rabanales Ctra. de Madrid km 396, 14071, Córdoba Universidad de Córdoba, España. Email: [email protected] RESUMEN Se plantea como objetivo un estudio comparativo del borde dorsal del cuello en equinos miniatura. Fueron estudiados un total de 125equinos miniatura de razas Falabella, MiniatureHorse, Pony Shetland, burro miniatura y enano (45 machos castrados y 80hembras), con edades comprendidas entre 2-20 años, sometidos a ejercicio de baja intensidad y diferentes condiciones de manejo y alimentación. Se realizó un estudio morfológico considerando la condición corporal, siguiendo el protocolo de adiposidad para la evaluación del borde dorsal del cuello descrito para equinos. El peso fue calculado empleando el sistema de ecuaciones KER descrito para caballos miniatura. Se realizó una estadística descriptiva a las medidas morfométricas del cuello y posteriormente se realizó un análisis de varianza (ANOVA). El estudio morfológico en evidenció una Puntuación 1.- 44% (55/125), Puntuación 2.- 32% (40/125), Puntuación 3.- 19% (24/125), Puntuación 4.- 5% (6/125). -

Michigan 4-H Miniature Horse Show Guidelines 2019
Michigan 4-H Miniature Horse Show Guidelines 2019 MSU is an affirmative-action, equal-opportunity employer. Michigan State University Extension programs and materials are open to all without regard to race, color, national origin, gender, gender identity, religion, age, height, weight, disability, political beliefs, sexual orientation, marital status, family status or veteran status. MI 4-H Miniature Horse Show Guidelines 2019 Table of Contents General Rules…………………………………………………………………………………………………………………………………….pg. 3 Halter………………………………………………………………………………………………………………………………………………..pg. 4 Color Class………………………………………………………………………………………………………………………………..……….pg. 4 Showmanship…………………………………………………………………………………………………………………………………….pg. 4 In-Hand Trail/Obstacle……………………………………………………………………………………….………………………………pg. 6 Liberty……………………………………………………………………………………………………………………………………………….pg. 6 Jumper In-Hand…………………………….…………………………………………………………………………………………………..pg. 6 Hunter In-Hand………………….………………………………………………………………………………………………………………pg. 7 Costume……………………………………………………………………………………………………………………………………….……pg. 9 General Driving Rules……………………………………………………………………………………………………………………….pg. 10 Pleasure Driving……………………………………………………………………………………………………………………………….pg. 10 Reinsmanship…………………………………………………………………………………………………………………..………………pg. 11 Versatility…………………………………………………………………………………………………………………………………………pg. 11 Ground Driven Obstacle………………………………………………………………………………………………….……………….pg. 12 Obstacle Driving……………………………………………………………………………………………………………….……………..pg. 12 Driven Dressage……………………………………………………………………………………………………………………………...pg.