Aromatic Electrophilic Substitution: The Arenium Ion Mechanism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Common Name: P-TOLUENE SULFONIC ACID HAZARD
Common Name: p-TOLUENE SULFONIC ACID CAS Number: 104-15-4 DOT Number: UN 2583 (solid, with more than 5% free Sulfuric Acid) UN 2585 (solid, with less than 5% free RTK Substance number: 1870 Sulfuric Acid) Date: October 1996 Revision: May 2003 ------------------------------------------------------------------------- ------------------------------------------------------------------------- HAZARD SUMMARY WORKPLACE EXPOSURE LIMITS * p-Toluene Sulfonic Acid can affect you when breathed in. No occupational exposure limits have been established for * p-Toluene Sulfonic Acid is a CORROSIVE CHEMICAL p-Toluene Sulfonic Acid. This does not mean that this and contact can cause severe skin and eye irritation and substance is not harmful. Safe work practices should always burns. be followed. * Exposure to p-Toluene Sulfonic Acid can irritate the nose, throat and lungs causing burning, dryness and WAYS OF REDUCING EXPOSURE coughing. * Where possible, enclose operations and use local exhaust ventilation at the site of chemical release. If local exhaust IDENTIFICATION ventilation or enclosure is not used, respirators should be p-Toluene Sulfonic Acid is a colorless, clear, or dark-colored worn. liquid or a colorless, crystalline (sand-like) material. It is used * Wear protective work clothing. to make dyes, drugs and other chemicals. * Wash thoroughly immediately after exposure to p- Toluene Sulfonic Acid and at the end of the workshift. REASON FOR CITATION * Post hazard and warning information in the work area. In * p-Toluene Sulfonic Acid is on the Hazardous Substance addition, as part of an ongoing education and training List because it is cited by DOT and NFPA. effort, communicate all information on the health and * This chemical is on the Special Health Hazard Substance safety hazards of p-Toluene Sulfonic Acid to potentially List because it is CORROSIVE. -

Structural Determination of Subsidiary Colors in Commercial Food Blue No
February 1998 7 Original Structural Determination of Subsidiary Colors in Commercial Food Blue No. 1 (Brilliant Blue FCF) Product (Received September 1, 1997) Hirosh1 MATSUFUJI*1, Takashi KUSAKA*1, Masatoshi TSUKUDA*1, Makoto CHINO*1, Yoshiaki KATO*2, Mikio NAKAMURA*2, Yukihiro GODA*3, Masatake TOYODA*3 and Mitsuharu TAKEDA*1 (*1College of Bioresource Sciences, Nihon University: 3-34-1, Shimouma, Setagaya-ku, Tokyo 145-0002, Japan; *2San-Ei Gen F. F. I., Inc.: 1-1-11, Sanwa-cho, Toyonaka, Osaka 561-0828, Japan; *3National Institute of Health Sciences (NIHS): 1-18-1, Kamiyoga, Setagaya-ku, Tokyo 158-8501, Japan) HPLC analysis revealed that five subsidiary colors were present in a commercial Food Blue No. l (Brilliant Blue FCF) product. Among them, major subsidiary colors C, D, and E were isolated. On the bases of spectroscopic analyses, their structures were identified as the disodium salt of 2-[[4-[N-ethyl-N-(3-sulfophenylmethyl)amino]phenyl][4-[N-ethyl-N-(4-sulfo- phenylmethyl)amino]phenyl]methylio]benzenesulfonic acid, the disodium salt of 2-[[4- [N-ethyl-N-(2-sulfophenylmethyl) amino]phenyl][4-[N-ethyl-N-(3-sulfophenylmethyl) amino]- phenyl]methylio]benzenesulfonic acid, and the sodium salt of 2-[[4-(N-ethylamino)phenyl][4- [N-ethyl-N-(3-sulfophenylmethyl)amino]phenyl]methylio]benzenesulfonic acid, respectively. Key words: Food Blue No. 1; Brilliant Blue FCF; FD & C Blue No. 1; subsidiary color; HPLC; coal-tar dye ally, it is the disodium salt of 2-[bis[4-[N-ethyl- Introduction N-(3-sulfophenylmethyl) amino] phenyl] meth- Twelve coal-tar dyes are presently permitted ylio]benzensulfonic acid [OSBA-(m-EBASA)(m- as food colors in Japan. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Intermediate Chemicals for Dyes Appendix to the Tariff
Harmonized Tariff Schedule of the United States (2004) Annotated for Statistical Reporting Purposes INTERMEDIATE CHEMICALS FOR DYES APPENDIX Harmonized Tariff Schedule of the United States (2004) Annotated for Statistical Reporting Purposes INTERMEDIATE CHEMICALS FOR DYES APPENDIX 2 This is supposed to be a blank page Harmonized Tariff Schedule of the United States (2004) Annotated for Statistical Reporting Purposes INTERMEDIATE CHEMICALS FOR DYES APPENDIX 3 This appendix enumerates those intermediate chemicals for dyes which are eligible for duty-free treatment under the provisions of general note 14 of the tariff schedule. Product CAS Number Acetaldehyde, (1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)- .............................................84-83-3 Acetamide, N-(3-amino-4-methoxyphenyl)- ...........................................................6375-47-9 Acetamide, N-(3-aminophenyl)- .....................................................................102-28-3 Acetamide, N-(4-aminophenyl)- .....................................................................122-80-5 Acetamide, N-(3-aminophenyl)-, monohydrochloride .....................................................621-35-2 Acetamide, N-(4-aminophenyl)-N-methyl- .............................................................119-63-1 Acetamide, N-(2,5-dimethoxy phenyl)- ...............................................................3467-59-2 Acetamide, N-(7-hydroxy-1-naphthalenyl- ............................................................6470-18-4 Acetamide, N-(2-methoxy-5-methyl -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Snoop Safety Data Sheet, New Zealand (SDS-SNOOP-NZ
Snoop ® Safety Data Sheet According to the Hazardous Substances and New Organisms Act (1996) Date of Issue: 06/10/2019 Version: 1.0 SECTION 1: IDENTIFICATION OF THE SUBSTANCE OR MIXTURE AND OF THE SUPPLIER 1.1. Product Name Product Form: Mixture Product Name: Snoop ® 1.2. Other Names Not available 1.3. Recommended Use Snoop® is a proprietary blend of water, non-ionic surfactants, and a bacterialcide. 1.4. Company Name, Address And Contact Details Company Distributor Swagelok Manufacturing Company, LLC Enter your contact information here 29495 F.A. Lennon Drive Solon, Ohio 44139 440-519-4000 www.swagelok.com 1.5. Emergency Phone Number Emergency Number : INFOTRAC: (800) 535-5035 SECTION 2: HAZARDS IDENTIFICATION 2.1. Classification Of The Substance Or Mixture GHS-NZ classification Not classified as a hazardous chemical. 2.2. GHS Label Elements, Including Precautionary Statements GHS-NZ Labeling No labelling applicable 2.3. Other hazards which do not result in classification Exposure may aggravate pre-existing eye, skin, or respiratory conditions. 2.4. Unknown Acute Toxicity (GHS-NZ) No data available SECTION 3: COMPOSITION/INFORMATION ON INGREDIENTS 3.1. Substance Not applicable 3.2. Mixture Name Synonyms Product Identifier % * GHS Ingredient Classification Water AQUA / Aqua (CAS-No.) 7732-18-5 > 99.44 Not classified Benzenesulfonic acid, mono- Benzenesulfonic acid, mono- (CAS-No.) 68649-00-3 0.255 Not classified C9-17-branched alkyl C9-17-branched alkyl derivatives, compounds with derivatives, isopropylamine 2-propanamine / C9-17 salts Branched alkylbenzenesulfonic acid, isopropylamine salt / Benzenesulfonic acid, mono- branched alkyl(C9-17) derivatives, compounds with 2-propanamine Dodecylbenzenesulfonic Benzenesulfonic acid, (CAS-No.) 26264-05-1 0.207 9.3C: Ecotoxicity to terrestrial acid, isopropylamine salt dodecyl-, compound with 2- vertebrates C, H433 propanamine (1:1) / Dodecylbenzenesulfonic acid 6.1D: Acute Tox. -

Lecture 13 Electrophilic Aromatic Substitution I 5.1 Principles
NPTEL – Chemistry – Principles of Organic Synthesis Lecture 13 Electrophilic Aromatic Substitution I 5.1 Principles The reaction occurs in two stages: the electrophile adds to one carbon atom of the aromatic ring, yielding a carbocation in which the positive charge is delocalized, and a proton is then eliminated from the adduct. H E H E H E E -H E 5.2 Formation of Carbon-Carbon Bonds 5.2.1 Friedel-Crafts Acylation Acylation of aromatic rings is generally peroformed using acid chloride or acid anhydride as an acylating agent in the presence of Lewis acid. O Z RCOCl, AlCl Z 3 R H2O Mechanism AlCl3 RCOCl RC=O + AlCl4 H H RC=O COR Z Z COR Z Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 26 NPTEL – Chemistry – Principles of Organic Synthesis In some circumstances, carboxylic acid is used as an acylating agent in the presence of a proton acid. HO OH O 2 PhOH, H2SO4 O O -H2O O O Phenolphthalein Indicator Intramolecular reactions are of particular value to construct cyclic systems. These reactions are usually carried out using dibasic acid anhydrides. For example, the synthesis -tetralone has been accomplished from benzene and succinic anhydride using AlCl3 in 80% yield. O O OH OH AlCl3 reduction + O O O O SOCl2 Cl AlCl3 O O Joint initiative of IITs and IISc – Funded by MHRD Page 2 of 26 NPTEL – Chemistry – Principles of Organic Synthesis Examples: 5 mol% Tb(OTf)3 CO H 2 PhCl O D.-M. Cui, C. Zhang, M. Kawamura, S. -

Alkylsulfonic Acids, Liquid
MATERIAL SAFETY DATA SHEET 1. IDENTIFICATION Product Name : ALKYLSULFONIC ACIDS, LIQUID Other Names : (C10-16) ALKYLBENZENESULFONIC ACID BENZENESULFONIC ACID, C10-16 ALKYL DERIVATIVES Uses : Feedstock for detergent derivative manufacture. Organisation Location Telephone Ask For 2 Swettenham Road Minto NSW 2566 02- Technical Redox Pty Ltd Australia 97333000 Officer 131126 Poisons Information Westmead NSW 1800- Centre 251525 2. HAZARD IDENTIFICATION Hazardous according to criteria of NOHSC CORROSIVE Risk Phrases R35 Causes severe burns. R22 Harmful if swallowed. Safety Phrases In case of contact with eyes, rinse immediately with plenty of water and seek S26 medical advice. S28:DOBENZ After contact with skin, wash immediately with plenty of water. S36/37/39 Wear suitable protective clothing, gloves and eye/face protection. ERMA New Zealand Approval Code : No Data HSNO Hazard Classification : No Data This Material Safety Data Sheet may not provide exhaustive guidance for all HSNO Controls assigned to this substance. The ERMA website www.ermanz.govt.nz should be consulted for a full list of triggered controls and cited regulations 3. COMPOSITION/INFORMATION ON INGREDIENTS Chemical Entity CAS No. Proportions (%) LINEAR ALKYL BENZENE SULPHONIC ACID [68584-22-5] 100 4. FIRST AID MEASURES Description of necessary measures according to routes of exposure Swallowed If swallowed, do NOT induce vomiting. Transport to nearest medical facility for treatment. If vomiting occurs naturally, keep head below hips to prevent aspiration. Eye Immediately flush eyes with large amounts of water holding eyelids open. Transport to the nearest medical facility for treatment. Skin Remove contaminated clothing. Immediately flush skin with plenty of water. Transport to the nearest medical facility for treatment. -
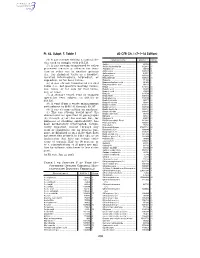
40 CFR Ch. I (7–1–14 Edition) Pt. 63, Subpt. F, Table 1
Pt. 63, Subpt. F, Table 1 40 CFR Ch. I (7–1–14 Edition) (4) A gas stream exiting a control de- Chemical name a CAS No. b Group vice used to comply with § 63.113. Aniline ................................................ 62533 I (5) A gas stream transferred to other Aniline hydrochloride ......................... 142041 III processes (on-site or off-site) for reac- Anisidine (o-) ..................................... 90040 II tion or other use in another process Anthracene ........................................ 120127 V (i.e., for chemical value as a product, Anthraquinone ................................... 84651 III Azobenzene ....................................... 103333 I isolated intermediate, byproduct, or Benzaldehyde .................................... 100527 III coproduct, or for heat value). Benzene ............................................ 71432 I (6) A gas stream transferred for fuel Benzenedisulfonic acid ...................... 98486 I Benzenesulfonic acid ........................ 98113 I value (i.e., net positive heating value), Benzil ................................................. 134816 III use, reuse, or for sale for fuel value, Benzilic acid ...................................... 76937 III use, or reuse. Benzoic acid ...................................... 65850 III Benzoin .............................................. 119539 III (7) A storage vessel vent or transfer Benzonitrile ........................................ 100470 III operation vent subject to § 63.119 or Benzophenone ................................. -

Samantha Leier
Chemoselective bioconjugation reactions of tyrosine residues for application in PET radiochemistry by Samantha Leier A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Cancer Sciences Department of Oncology University of Alberta © Samantha Leier, 2018 ABSTRACT Achieving chemoselectivity while maintaining bioorthogonality are among some of the major challenges in bioconjugation chemistry. Bioconjugation chemistry has important applications in PET radiochemistry. While modern bioconjugation techniques rely predominantly on lysine and cysteine residues as targets for bioconjugation, other bioconjugation techniques that selectively target other amino acids would contribute greatly to the development of new PET radiotracers. Although only modestly prevalent and often buried within the protein structure, tyrosine represents an attractive target for bioconjugation. Tyrosine residues can be selectively targeted by both luminol derivatives and aryl diazonium salts. Luminol derivatives react via an ene-like reaction where as diazonium compounds react via electrophilic aromatic substitution to produce a stable conjugate. Recently, luminol derivatives have been reported for the introduction of fluorescent probes into proteins and diazonium salts have been reported for the introduction of radiometals into tyrosinamide-containing polymers. To the best of our knowledge, neither technique has been applied to PET radiotracer synthesis. Therefore, the aim of this project is the chemoselective introduction of radionuclides onto tyrosine residues under mild conditions. Specifically, we describe the preparation of 18F-labeled luminol derivatives as well as 64Cu- and 68Ga-labeled diazonium salts as novel building blocks for subsequent coupling with tyrosine residues under mild conditions. Luminol derivative N-(4-[18F]fluorobenzyl)-2-methyl-1,4-dioxo- 1,2,3,4-tetrahydro-phthalazine-6-carboxamide (29) was synthesized in 48% radiochemical yield. -

Coupling of Methanol and Carbon Monoxide Over H‐ZSM‐5 to Form
Angewandte Communications Chemie International Edition:DOI:10.1002/anie.201807814 Heterogeneous Catalysis Hot Paper German Edition:DOI:10.1002/ange.201807814 Coupling of Methanol and Carbon Monoxide over H-ZSM-5 to Form Aromatics Zhiyang Chen, Youming Ni, Yuchun Zhi, Fuli Wen, Ziqiao Zhou, Yingxu Wei, Wenliang Zhu,* and Zhongmin Liu* Abstract: The conversion of methanol into aromatics over via hydrogen transfer.[3] Although modifications with metals unmodified H-ZSM-5 zeoliteisgenerally not high because the such as Zn,[2a,c,e] Ga,[2d] and Ag[2b] increase aromatics forma- hydrogen transfer reaction results in alkane formation. Now tion by enhancing the dehydrogenation of alkanes via circa 80%aromatics selectivity for the coupling reaction of catalysis by these metal species,[4] some methanol decompo- methanol and carbon monoxide over H-ZSM-5 is reported. sition and the formation of unrecoverable catalyst structure Carbonyl compounds and methyl-2-cyclopenten-1-ones owing to metal evaporation, segregation, and aggregation are (MCPOs), which were detected in the products and catalysts, inevitable.[2a,5] respectively,are considered as intermediates.The latter species Recently,Cheng et al. reported that aromatization could 13 can be synthesized from the former species and olefins. C be enhanced by CO over the bifunctional ZnZrOx/H-ZSM-5 isotope tracing and 13Cliquid-state NMR results confirmed that catalysts in the conversion of syngas or intermediate meth- the carbon atoms of CO molecules were incorporated into anol because CO plays arole in the removal of Hspecies and MCPOs and aromatic rings.Anew aromatization mechanism subsequent formation of methanol. As aresult, it can be that involves the formation of the aboveintermediates and co- deduced that ZnZrOx is indispensable to the catalysis occurs with adramatically decreased hydrogen transfer mechanism.[6] reaction is proposed. -
![A New Rhodium Catalyst: Formation of [Rh(CO)4]+ in Concentrated](https://docslib.b-cdn.net/cover/9875/a-new-rhodium-catalyst-formation-of-rh-co-4-in-concentrated-2179875.webp)
A New Rhodium Catalyst: Formation of [Rh(CO)4]+ in Concentrated
1540 J. Org. Chem. 2000, 65, 1540-1543 Notes 6-8 A New Rhodium Catalyst: Formation of under an atmospheric pressure of CO. [Pt(CO4)]- + [Rh(CO)4] in Concentrated Sulfuric Acid [Sb2F11]2 has recently been used for the stereospecific and Its Application to Carbonylation of tetramerization of 2-propynol and the polymerization of Olefins arylacetylenes.9 Rhodium catalysts, most of which work in organic solvents, have been employed in many important reac- Qiang Xu,* Hisako Nakatani, and Yoshie Souma tions, such as the hydrogenation,10 hydroformylation, and 1 Osaka National Research Institute, AIST, MITI, 1-8-31, carbonylation of unsaturated compounds. In this paper, Midorigaoka, Ikeda, Osaka, 563-8577, Japan we report a new rhodium carbonyl catalyst in concd H2SO4, with which olefins react with CO to produce Received October 21, 1999 tertiary carboxylic acids in high yields at atmospheric pressure and room temperature. This work extends the family of the cationic metal carbonyl catalysts for car- Introduction bonylation of olefins from groups 11 and 10 to the group Metal carbonyls have played a very important role in 9 elements. chemistry and the chemical industry.1 For the typical metal carbonyls such as Ni(CO)4,Co2(CO)8, Fe(CO)5, and Results and Discussion Mn(CO) 3-, the average vibrational frequencies, ν(CO), 4 Formation of Rhodium(I) Tetracarbonyl Cation, are considerably lower than the value for free CO, 2143 + cm-1, mainly due to the metal-to-CO π-back-bonding.2 [Rh(CO)4] , in Concentrated H2SO4. Very recently, new Rh(I) and Rh(III) carbonyl cations have been gener- Reactions catalyzed by such metal carbonyls usually + require drastic conditions; for example, the Roelen and ated.